INTRODUCTION
Direct bilirubinemia is defined as an increase in total bilirubin from Ōēż5 to Ōēź1 mg/dL or a direct fraction Ōēź20% if total bilirubin is >5 mg/dL, which are the accepted standards for clinical cholestasis [1]. In neonates, genetic disorders of primary biliary and cellular function and metabolic diseases must be differentiated, and secondary liver injury from various causes must be considered, including abnormal immune responses, hemolysis, hemodynamic changes, and phytosterol accumulation from parenteral nutrition [2,3]. In addition, the functionally immature liver of a neonate may respond to changes in systemic status during the perinatal period, causing nonspecific hepatocellular injury and subsequent cholestasis. Cholestasis of unknown etiology is reported in 13% to 26% of newborns [4]. Owing to a complex etiologic background and limited technical resources, diagnostic planning for cholestasis that persists after biliary atresia (BA) differentiation remains a challenging clinical problem.
In cases of persistent cholestasis, the presence of hemolytic disease should be evaluated, as the occurrence of cholestasis and its progression to chronic liver disease due to neonatal hemolytic disease have been demonstrated in some population-based studies [5]. However, the mechanisms of liver pathology that account for this as well as clinical guidelines are unclear. This is attributed to nonspecific hematological and biochemical changes associated with comorbidities in the perinatal circulatory and respiratory systems, making the assessment of specific hemolytic diseases difficult.
Herein, we describe the identification of extensive hemosiderosis due to hemolysis in the course of persistent cholestasis of unknown etiology in a neonate with a history of respiratory circulatory system complications, the diagnosis of ╬▓-thalassemia as the underlying cause of persistent cholestasis, and the course of spontaneous recovery of liver pathology.
CASE REPORT
The female patient was born at 38 weeks and 2 days of gestation, with a birth weight of 3,380 g via a repeated elective Cesarean section at another hospital and was transferred to Ajou University Hospital because of respiratory distress immediately after birth. No complications during pregnancy or labor and no significant maternal history were recorded. Both parents are Korean, the patient is the second child, and no specific family history of the disease was reported. Upon arrival at the hospital, her body temperature, blood pressure, heart rate, respiratory rate, and oxygen saturation on room air were 36.5 ┬░C, 64/38 mm Hg, 158 beats/min, 51 breaths/min, and 50%, respectively. The patient was assessed for acute respiratory distress syndrome using chest radiography; hence, a surfactant was administered. Echocardiography was performed for suspected persistent pulmonary arterial hypertension with a >20% difference in upper (right hand) and lower (right foot) oxygen saturation without improvement. The echocardiogram revealed a large 5.3- mm patent ductus arteriosus (PDA), a D-shaped left ventricle, and severe pulmonary hypertension with no improvement in symptoms after the administration of inotropics (dopamine, dobutamine, epinephrine, and norepinephrine), milrinone, and nitric oxide inhalation.
On day 2 of admission, she developed a 39 ┬░C fever, and her condition progressed to sepsis with disseminated intravascular coagulation in blood tests. Subsequently, the patient was administered broad spectrum antibiotics (ampicillin/sulbactam 100 mg/kg and cefotaxime 50 mg/kg twice daily) and immunoglobulin 0.5 mg/kg for 2 days. Her vital signs began to stabilize on day 6 of admission. PDA ligation was performed on the 21st day of hospitalization, and the patientŌĆÖs respiration stabilized postoperatively. Thereafter, the patientŌĆÖs saturation remained stable without requiring additional oxygenation.
Direct bilirubinemia (6.7 mg/dL) was first detected on day 4 of hospitalization, presumably because of hemodynamic instability due to cardiorespiratory compromise and prolonged total parenteral nutrition. However, direct bilirubinemia (11.9 mg/dL) persisted despite improvement in the respiratory status. Abdominal sonography performed to evaluate the persistent cholestasis revealed a negative triangular cord sign, no apparent findings in the gallbladder (GB), and no findings suggestive of inspissated bile syndrome. Conservative care such as early trophic feeding, cyclic total parenteral nutrition, and other conservative measures were taken; however, direct bilirubinemia (14.7 mg/dL) persisted despite stabilization of the patient's enteral feeding regimen at approximately 24 days of age.
The color of the patientŌĆÖs fecal matter also changed to pale yellow. A hepatobiliary scan was also performed, which indicated decreased hepatic extraction and delayed parenchymal transit times. The GB and intestines were not visualized until 24 hours, and suspected BA was confirmed. Liver biopsy was performed to confirm the diagnosis. Mild portal inflammation with moderate lobular activity was observed with multinucleated hepatocytes, hepatocellular swelling, and severe cholestasis with a canalicular pattern. The initial biopsy confirmed a high probability of neonatal hepatitis. Considering the patientŌĆÖs relatively low gammaglutamyl transferase levels with a rather nonspecific neonatal hepatitis impression, a gene test (ABCB11 or AT8B1) was performed to rule out progressive familial intrahepatic cholestasis.
Blood transfusions were administered following our neonatal practice guidelines, with a total of eight transfusions provided to sustain a hematocrit (Hct) level between 50% and 55% for infants diagnosed with persistent pulmonary hypertension of the newborn.
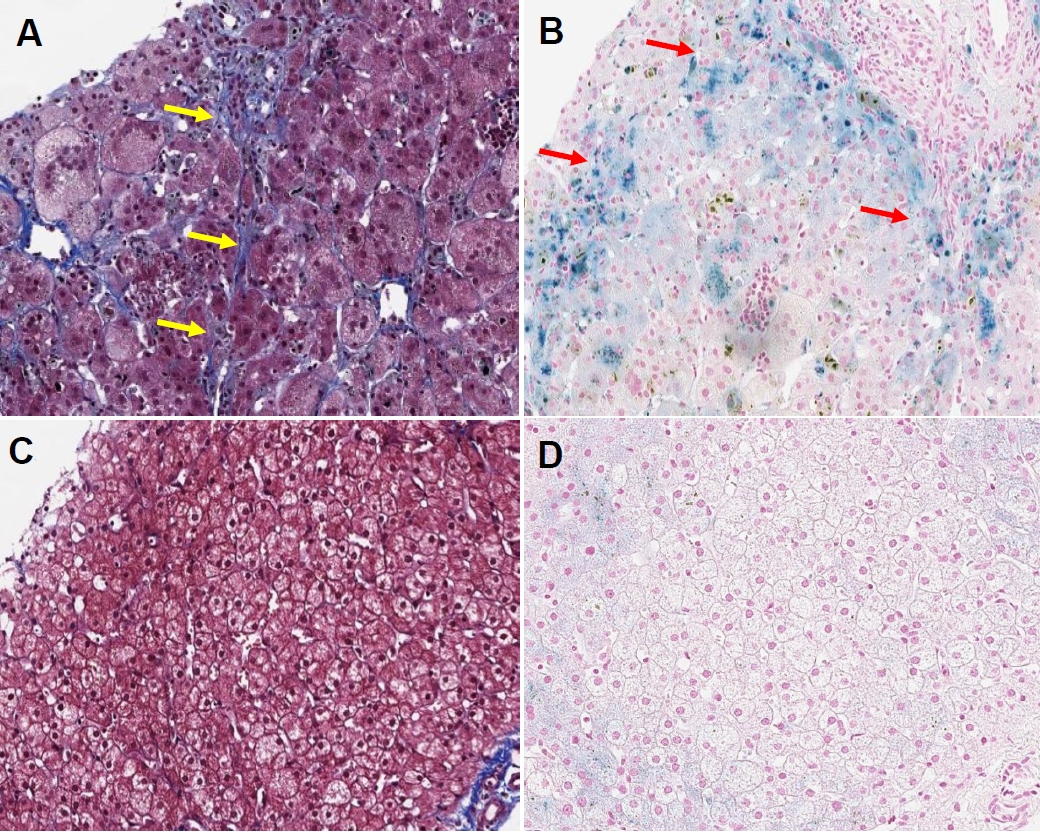
From approximately 2 months after the first liver biopsy, a second liver biopsy was performed, which revealed persistent cholestasis with giant cell transformation and diffuse hemosiderin deposition (Figure 1). Magnetic resonance imaging (MRI) and buccal biopsy were performed because of persistently high ferritin levels and liver biopsy findings, despite the typical clinical course of gestational alloimmune liver disease (GALD). Although the MRI confirmed secondary hemochromatosis, the buccal biopsy result was negative.
Blood transfusions were administered according to the established neonatal practice guidelines. These transfusions were predicted based on the necessity for moderate mechanical ventilation, characterized by parameters such as a mean airway pressure (MAP) exceeding 8 cmH2O for conventional mechanical ventilation (CMV), a MAP of >14 cmH2O for high-frequency oscillatory ventilation (HFO), or a fraction of inspired oxygen (FiO2) of >0.4, concomitant with an Hct of <30% and hemoglobin (Hb) level of <10 g/dL. In instances where minimal mechanical ventilation was indicated, interventions such as continuous positive airway pressure, CMV with a MAP of <8 cmH2O, or HFO with a MAP of <14 cmH2O, or FiO2 <0.4, and transfusions were considered if the Hct decreased to <25% and Hb was <8 g/dL. Moreover, when Hct decreased to <20% and Hb decreased to <7 g/dL, an oxygen therapy regimen was implemented exclusively, precluding the need for mechanical ventilation. This approach was similarly applicable in cases of sustained tachycardia or tachypnea persisting >24 hours or a two-fold elevation in oxygen requisites within a 48-hour timeframe, instances of metabolic acidosis, or significant surgical interventions. When Hct and Hb decreased to <18% and <6 g/dL, respectively, blood transfusion was performed without conditional constraints.
Concurrently, karyotyping was normal (46, XX), a microarray revealed a 2.7 Mb deletion at 11p15.4, which was identified as a ╬▓-globin gene cluster including the HBB, HBD, HBE1, HBG1, and HBG2 genes. These genes are associated with heart disease and Hb genes and red blood cell abnormalities [6]. Therefore, assuming that hemolytic cholestasis was caused by hemoglobinopathy, the 18 thalassemia battery for thalassemia, ╬▓-thalassemia, and HBB gene confirmation tests were performed, thus subsequently confirming ╬▓-thalassemia.
The patient receiving no special medications demonstrated improvement in bile acid-dependent laboratory tests, with a normal range of direct bilirubin levels at 23 weeks of age (Figure 2). Transaminitis also improved. Since then, the patient has had a stable course with normal growth and development, with only folic acid supplementation and without requiring transfusions. A third liver biopsy performed at approximately 1 year of age revealed an intact lobular architecture with multifocal swollen hepatocytes and mild hemosiderin deposition in the periportal areas (Figure 1), confirming overall improvement. Outpatient follow-up indicated stable blood work results.
DISCUSSION
╬▓-Thalassemia is a genetic disorder characterized by reduced or defective synthesis of the ╬▓-globin chain in the Hb. It is caused by a point mutation or, more rarely, a deletion in the ╬▓-globin gene on chromosome 11, resulting in reduced (╬▓+) or absent (╬▓0) synthesis of the ╬▓-chain of Hb. This results in inefficient red blood cell production owing to an imbalance in the alpha-globin chain. The clinical presentation is broadly divided into severe, moderate, and mild thalassemia, with individual differences ranging from severe to asymptomatic anemia. The total annual incidence of symptomatic individuals is approximately 1 in 100,000 people worldwide and 1 in 10,000 people in the European Union. In Korea, the incidence rate is 0.41 in 100,000 people and is gradually increasing owing to the impact of increasing immigration [7,8]. A single institutional report from Korea has identified nine pediatric cases over an 11-year period, of which eight were ╬▓-thalassemia besides one alpha thalassemia. The median age at diagnosis is 4.3 years, and 78% of cases have an asymptomatic, mild phenotype [9]. In newborns, it can be detected early in life as Coombs-negative hemolytic anemia with hepatosplenomegaly and direct or indirect hyperbilirubinemia due to hemolysis [10].
Thalassemia is diagnosed based on hematological and molecular tests. Microarrays have been reported to have screening utility for patients with thalassemia in Asia [11]. In the present case, the patient received a definitive diagnosis of the DNA genotype after identification of a large deletion by microarray. Hence, microarrays may be a safe and rapid screening evaluation tool for primary inherited erythrocyte diseases, including ╬▓-thalassemia, in the early stages of a complex clinical situation.
Neonatal hemolysis is classified as an extracellular (acquired) or intrinsic (genetic) red blood cell defect. An overload of iron components due to hemolysis can cause hemosiderosis in the tissues. The severity of cholestasis due to primary erythrocyte disease (hemoglobinopathy) can be observed in sickle cell hepatopathy (SCH), which results in vascular obstructive disorders with hemosiderosis and cholelithiasis of the liver tissue due to intravascular iron overload [12]. However, reports on neonatal cases and natural history are scarce [13]. The histological characteristics of SCH are characterized by Cooper cell erythrocytosis, hemosiderosis, sinusoidal dilatation, and intra-sinusoidal sickling [14]. In this case, liver pathology suggested hemosiderin accumulation (siderosis) on two examinations, progression of extramedullary hematopoiesis, perisinusoidal fibrosis, and giant cell formation. It was differentiated from SCH as it was not characterized as a vaso-occlusive disorder but rather exhibited characteristics closer to the pathology of GALD; however, typical GALD levels of necrosis were not observed [15].
GALD has a high fatality rate and requires aggressive intravenous immunoglobulin (IVIG) and plasmapheresis in preparation for liver transplantation and as bridge therapy. Considering the opportunity cost to the patient and burden on technical resources, an urgent exclusionary diagnosis is essential. The persistence of hyperferritinemia on biochemical testing required differentiation from GALD, as did the similarities identified in the histological findings. Although GALD can develop in the early perinatal period and this case was outside the typical timing of GALD onset, we excluded the possibility that the IVIG administration for treating sepsis early in hospitalization may have caused a partial atypical response. Ultimately, the less acute progression of the liver dysfunction, buccal biopsy results, and MRI findings confirmed the exclusion of this diagnosis. The reported positive rate of buccal salivary biopsy in GALD by Smith et al. [16] was 85.7%; however, the biopsy in this case was negative. MRI confirmed the difference in primary/secondary iron deposition characteristics with and without pancreatic sparing, which accords with the conclusions of a previous imaging report [17].
The reversibility of advanced cirrhotic liver changes is difficult to predict in the early stages. Outpatient follow-up and a 1-year biopsy demonstrated a level of biochemical and histological recovery that did not require separate chronic liver disease management beyond the management of primary hemoglobinopathy. Mechanistic explanations of cholestasis in hemolytic diseases and its subsequent progression to cirrhosis are lacking. Teng et al. [5] have reported that in a population-based survey in Northern Europe, the rate of cholestasis in patients with neonatal hemolytic anemia was 7% (11/149) and did not progress to chronic liver disease at 2 years of age. In addition, a study on adult transfusion-dependent patients with ╬▓-thalassemia has reported that >70% of patients had cirrhotic liver changes of stage 3 or higher, and the extent of this fibrosis was related to age [18]. A good natural history may be expected in cases of hemolytic cholestasis and that an early and appropriate diagnosis is important for establishing conservative strategies to minimize iron overload.
To the best of our knowledge, this is the first case report in Korea to evaluate the possibility of progression to cholestasis and cirrhosis due to the hemolytic burden caused by early postnatal hemoglobinopathy. We hope that this case report would encourage further research on the necessity and feasibility of differential diagnosis of hemoglobinopathy-induced hemolysis as a secondary evaluation in patients with neonatal cholestasis.