INTRODUCTION
Cyanosis during the neonatal period is often concerning for neonatologists. Although the most common causes of central cyanosis are respiratory diseases and cyanotic congenital heart diseases, cyanosis can also occur in methemoglobinemia. Methemoglobin (MetHb) is produced when the iron in the hemoglobin molecule is in the oxidized ferric state instead of the ferrous state [1]. MetHb, unlike its counterpart oxyhemoglobin, has a higher affinity for oxygen than that for carbon dioxide [2]. It should be considered in the differential diagnosis of cyanosis in patients with normal cardiovascular and pulmonary functions.
Methemoglobinemia in neonates may be congenital (due to hereditary deficiency of nicotinamide adenine dinucleotide [NADH] cytochrome b5-reductase enzyme) or acquired [3]. Acquired methemoglobinemia in newborns can be due to endogenous conditions, such as diarrhea, sepsis, and acidosis, exogenous drugs, or toxins [4].
Methemoglobinemia is more common in infants and children because fetal hemoglobin (HbF) is more easily oxidized to metHb [1]. Lower levels of NADH-MetHb reductase and lower glutathione peroxidase activity in infants and children than in adults also contribute to their higher predisposition to developing methemoglobinemia [1]. HbF variants causing methemoglobinemia are rare causes of neonatal cyanosis, which can cause transient cyanoReceived sis lasting for almost 6 months [1]. Here, we present a case report and literature review of a newborn with transient cyanosis due to mutations in the gamma chain of HbF.
CASE REPORT
A 2.73 kg female infant was born to a primiparous mother at 37 weeks and 3 days of gestation at a local hospital. The baby was vigorous at birth but was mildly cyanosed, and her oxygen saturation was 80% at room air. No respiratory distress was observed. The baby was administered oxygen, fraction of inspired oxygen (FiO2)ŌĆō1.0, which did not have the desired effect, and her oxygen saturation remained between 80% and 85%. Systemic examination, complete blood count, and routine biochemistry were unremarkable. Chest radiography revealed well-expanded lungs with normal lung parenchyma. Echocardiography (ECHO) revealed a dilated right atrium and ventricle and a small atrial septal defect. There was mild tricuspid regurgitation with a peak gradient of 55 mm Hg and mild flow acceleration in the right pulmonary artery with no significant gradient. Her arterial blood sample was dark red in color, and arterial blood gas analysis showed a pH 7.403, partial pressure of carbon dioxide (pCO2) 31.9 mm Hg, and partial pressure of oxygen (pO2) 102 mm Hg; the oxygen saturation in blood gases was 98%, but simultaneous pulse oximetry showed a saturation of only 86%. All MetHb values were >7%. Repeat samples obtained on day 3 showed persistently elevated MetHb levels of 6.8%.
Therefore, she was transferred to our center for further evaluation and care. At admission, on day 5 of life, the baby was active and vigorous with no signs of respiratory distress. She had mild cyanosis around her eyelids, and all vital signs were within normal limits. The oxygen saturation at room air was 85% to 88%. Head-to-toe examination was normal, and there were no dysmorphic features. The systemic evaluation was normal. Chest radiography revealed normal lungs and cardiac silhouettes. A hyperoxia test was performed, and arterial blood gas of blood taken from the right radial artery showed a pH 7.41, pCO2 31 mm Hg, pO2 127 mm Hg, HCO3 21.2 mEq/L, and MetHb 7.2%. Her oxygen saturation level was maintained at 88%. A bedside ECHO performed on admission revealed a suspicious drop out in the interventricular septum with a small atrial septal defect measuring 4 mm, moderate tricuspid regurgitation, and findings consistent with mild-to-moderate pulmonary hypertension. We made a provisional diagnosis of methemoglobinemia, and a trial of hood-box oxygen therapy was initiated, which marginally increased the oxygen saturation to a maximum of 90%. The oxygen saturations of both parents were normal, as were their MetHb levels.
In light of the clinical and laboratory findings, the maternal medical, family, and social histories were revisited. The mother and remaining household members were asymptomatic, ruling out the environment, food, or water sources as a possible etiology. Water sample analysis revealed normal nitrate levels. The motherŌĆÖs antenatal history revealed that she was prescribed L-arginine granules (each 5 g sachet contained 3 g L-arginine), one sachet daily and controlled release nitroglycerin tablets, two tablets (5.6 mg) in the morning, and one tablet (2.6 mg) at night for oligohydramnios and fetal growth restriction. However, during the month prior to delivery, she was being administered 5.6 mg nitroglycerine tablets twice daily. These drugs, either alone or in combination, can contribute to methemoglobinemia. However, this was ruled out as an etiology because the motherŌĆÖs oxygen saturation and MetHb levels were normal at room air. Because the MetHb levels revealed mild disease, methylene blue therapy was considered while awaiting results on the glucose-6-phosphate dehydrogenase (G6PD) levels in the baby; however, this was deferred because some studies suggest an increased risk of hemolysis with methylene blue therapy in patients with G6PD deficiency [5]. Considering the working diagnosis of congenital methemoglobinemia and to enable normal growth and development, we initiated ascorbic acid therapy at a modest dose of 40 mg once daily. We continued to monitor her oxygen saturation and MetHb levels for the next few days. She showed a gradual improvement in oxygen saturation, and oxygen could be weaned off by day 10. She was discharged on the 12th day of life with a MetHb level of 6.6% and was advised to continue ascorbic acid therapy, with her saturations predominantly between 90% and 92% at room air. At follow-up on day 21, she thrived and maintained an oxygen saturation of 90%. Her MetHb level reduced gradually and at the 1-month postdischarge follow-up, it was 5.8%. A 3-month follow-up showed a MetHb level of 3.3%. Whole exome sequencing revealed a heterozygous missense variation in exon 2 of the hemoglobin subunit gamma 2 (HBG2) gene, which resulted in the amino acid substitution of tyrosine for histidine at codon 93, consistent with fetal hemoglobin molecule to methemoglobin (Hb-FM) Fort Ripley, a gamma chain variant hemoglobinopathy. This causes reduced affinity for oxygen due to steric inhibition of oxygen-binding and/or increased oxidation of the Hb-FM, which has decreased oxygen-binding capacity. Segregation analysis showed that the asymptomatic father was a heterozygous carrier of the mutation.
DISCUSSION
MetHb is formed when the deoxygenated heme molecule is oxidized from the ferrous to the ferric state. MetHb has a reduced ability to transport oxygen, and thus produces functional anemia [1,2]. In addition, the ferric heme group causes allosteric changes to the hemoglobin molecule, which makes the ferrous heme moieties on the same hemoglobin tetramer bind oxygen more tightly. This is known as the Darling-Roughton effect [6]. It is estimated that approximately 3% of hemoglobin is oxidized to MetHb daily [7]. Methemoglobinemia may also be induced by several oxidizing agents, such as nitroglycerin, sulfonamides, phenytoin, and local anesthetics, including lidocaine and prilocaine [3,4].
HbF is produced from the 6th week of gestation and switches to the major adult hemoglobin A (HbA) and minor adult hemoglobin subunit alpha 2 (HbA2) tetramers immediately before birth [8]. As a result of a second switch at 6 months of age, HbF accounts for less than 5% of the total hemoglobin. Hence, the manifestations due to the abnormal gamma chain disappear within this time-frame [9]. The majority of discovered hemoglobin M (HbM) and Hb-FM variants are caused by the substitution of the distal or proximal histidine residues with tyrosine, so that the heme iron is stabilized in the Fe3+ form [10]. Patients with HbM disease usually have MetHb levels between 15% and 30% and remain asymptomatic [3]. The transmission of HbM is autosomal dominant, and life expectancy of affected patients remains unchanged [8]. Neonatal cyanosis associated with Hb-FM variants is rare. Only six gamma chain variants have been identified in the literature (Hb-FM Osaka, Hb-FM Fort Ripley, Hb-FM Circleville, HbF-Cincinnati, Hb-FM Toms River, and Hb Viseu) [8,9]. The results of the literature review of published case reports of gamma chain variants are summarized below (Table 1) [9-16].
A simple and quick bedside test for methemoglobinemia involves placing two drops of blood on a white filter paper and evaluating the color change upon exposure to oxygen. The chocolate -brown appearance of MetHb does not change with time; however, deoxyhemoglobin initially appears dark red and then brightens after exposure to atmospheric oxygen [14]. Next-generation sequencing may be the most efficient approach for diagnosing fetal hemoglobinopathy when clinical data support the diagnosis [15].
The treatment of methemoglobinemia should be guided by the etiology, severity, and blood levels of MetHb. Metabolic acidosis may be present depending on the severity and duration of tissue hypoxia. Basic first aid management should be carried out, including supplemental oxygen (100%), ensuring airway patency, and hemodynamic support [16]. In asymptomatic or mild cases, only close observation may be required. Congenital methemoglobinemia requires genetic counselling and explanation of the long-term prognosis to the parents [17]. If an acquired etiology is confirmed, methemoglobinemia should usually resolve within 36 hours of removing the offending agent due to the action of physiological reducing mechanisms [1,17].
Methylene blue is an antidote for methemoglobinemia. It acts as a cofactor and accelerates the nicotinamide adenine dinucleotide phosphate-dependent MetHb reductase system, which reduces methylene blue to methylene leucoblue [17]. It has limited effectiveness in patients with HbM, G6PD deficiency, and neurologic abnormalities [17,18]. Ascorbic acid (vitamin C) directly reduces MetHb, rather than restoring the normal enzymatic reduction mechanism. Ascorbic acid at doses of 200 to 500 mg/day in three to four divided doses have been attempted, but resolution has been noted to be slow [19]. We used much lower doses (40 to 50 mg/day, equivalent to the recommended daily allowance for babies aged 6 to 12 months) in this infant due to lack of strong evidence of its benefit and clear advice in various neonatal formularies regarding the safe therapeutic dose. Hyperbaric oxygen therapy and exchange transfusion are other treatment modalities that have been tried [20].
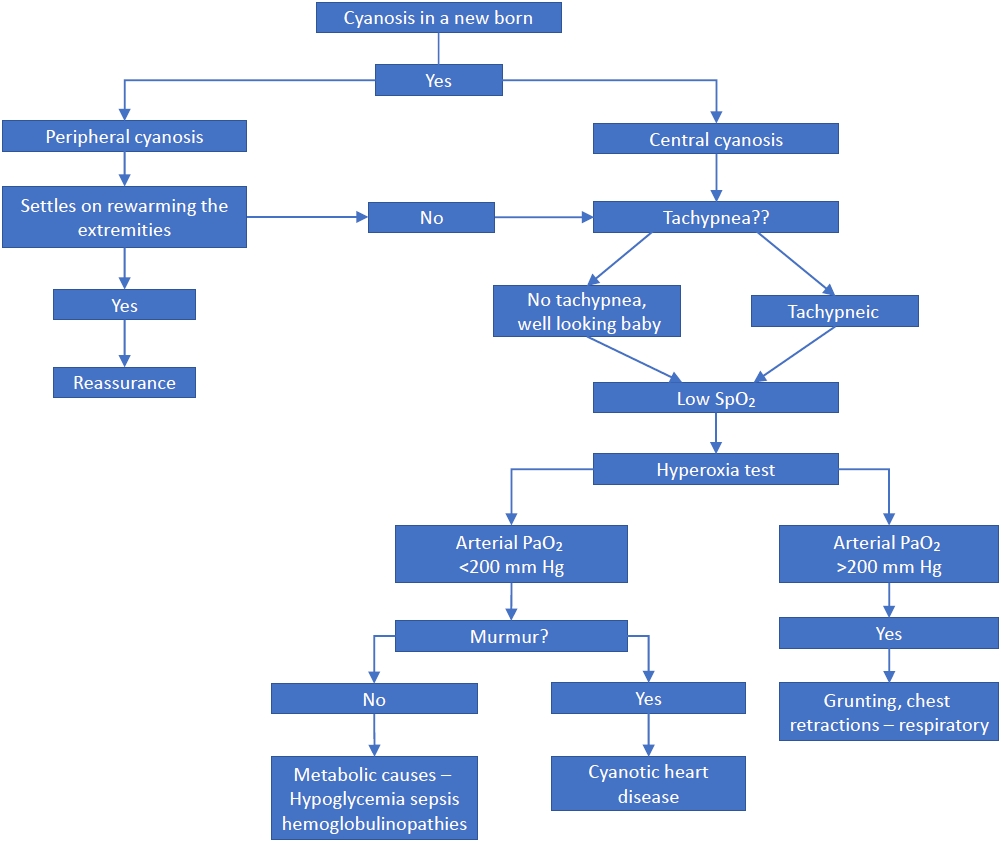
This case report highlights a rare cause of neonatal cyanosis that should be considered in the differential diagnosis of a cyanotic newborn, especially when more common causes are excluded. A pragmatic approach for evaluating cyanosis in a newborn is shown in Figure 1. The benefits of invasive procedures or tests should be carefully evaluated in a cyanotic baby who appears healthy with modestly elevated MetHb, normal arterial pO2, and no signs of cardiopulmonary disease. Some of the Hb variants have normal electrophoresis results, and the diagnosis is made by exome sequencing [15].
In conclusion, methemoglobinemia is a rare cause of hypoxia and cyanosis, which can be potentially fatal if not addressed in a timely manner or left untreated. Genetic evaluation is advisable in infants with unexplained methemoglobinemia. Early diagnosis of methemoglobinemia due to gamma chain mutation in HbF, as in our case, helps to reassure parents and prevent unnecessary aggressive investigations and treatments. Those with mutations in the gamma chain of HbF have an excellent prognosis, and their symptoms and the elevated MetHb levels normalize over 4 to 6 months of life.