INTRODUCTION
Periventricular nodular heterotopia (PNH) is a cortical development malformation caused by disruption of neuronal migration, resulting in ectopic neuronal nodules along the lateral ventricles of the brain [1]. PNH is usually diagnosed based on brain imaging studies, typically magnetic resonance imaging (MRI), which is usually performed when the patient develops epilepsy, the main presenting symptom of PNH. For this reason, PNH is usually diagnosed in adolescence unless it is associated with other congenital anomalies [2]. The most common causative gene is filamin A (FLNA), which is located on the X chromosome [3]. A less common form is mutation of the adenosine diphosphate-ribosylation factor guanine exchange factor 2 (ARFGEF2) gene, which is inherited in an autosomal recessive pattern [3].
We report a case of a newborn with unilateral PNH accompanied by symptomatic patent ductus arteriosus (PDA), persistent feeding cyanosis, and recurrent apnea.
CASE REPORT
A female infant was born at 34 weeks and 1 day of gestation via planned cesarean section. No abnormal signs were detected on fetal ultrasonography. At birth, the body weight was 2,250 g (59 percentile), body height was 43 cm (33 percentile), and head circumference was 31.5 cm (69 percentile). The Apgar scores were 7 and 9 at 1 and 5 minutes, respectively. The infant’s parents were 41 years of age at the time of delivery, and her mother had a medical history of preeclampsia and type 2 diabetes mellitus on insulin treatment, which was diagnosed 10 years ago. The patient’s mother was a gravida 1, para 0, abortus 1 woman with a history of spontaneous abortion at 6 weeks of gestation. The patient was the first child with no familial history of neurological disorders such as epilepsy, cerebral palsy, developmental delay, or hearing loss.
She was born by cesarean section at Ajou University Hospital because of uncontrolled maternal preeclampsia and was admitted to the neonatal intensive care unit because of prematurity and low birth weight. Her initial blood pressure was 52/25 mm Hg, body temperature was 36.5°C, pulse rate was 158 beats/min, respiratory rate was 29 breaths/min, and O2 saturation was 90%. The initial physical examination revealed no abnormal findings, except for mild tachypnea and peripheral cyanosis. Following the diagnosis of transient tachypnea of the newborn, 25% oxygen was administered with a high-flow nasal cannula at 7 L/min. Her initial blood glucose, serum calcium, and serum ionized calcium levels were 18, 6.5, and 3.3 mg/dL respectively. She was supplemented with 10% dextrose (2 mL/kg) and intravenous calcium gluconate. Serum glucose levels recovered to 60 mg/dL. Other serological test results were all within normal ranges. On the 2nd day of life, total parenteral nutrition (TPN) was initiated owing to poor feeding, frequent vomiting, and recurrent hypoglycemia with a serum glucose level of 20 mg/dL. Intravenous 10% dextrose (2 mL/kg) was administered and the serum glucose level recovered to 71 mg/dL. TPN was initiated at a glucose infusion rate of 3 mg/kg/min; calcium was not supplemented, since hypocalcemia was resolved to 7.9 mg/dL. The serum glucose level normalized to 80 mg/dL on the 3rd day, and the hypoglycemia did not recur. On the 5th day, echocardiography revealed a 3.4-mm PDA with a left-to-right shunt. The patient still needed a high-flow nasal cannula with 21% oxygen at a rate of 5 L/min to breathe, and on the 9th day we had to apply a mechanical ventilator due to signs of symptomatic PDA, including worsening respiratory distress, tachypnea of 60 breaths/min, and severe chest retraction. Mechanical ventilation was initiated in synchronized intermittent mandatory ventilation mode and set on a 23% fraction of inspired O2 and a positive end-expiratory pressure of 5 mm Hg. On the 11th day, the PDA was surgically ligated. She showed no signs of coagulopathy, and serum prothrombin and activated partial thromboplastin times were normal. The patient showed improvement in respiration, and the ventilator was weaned the day after the surgery.
She had feeding problems since birth, cyanosis during bottle-feeding, and a significant amount of milk residue. On the 16th day, TPN was removed and bottle-feeding was performed in combination with gavage tube feeding. She also suffered from frequent apnea; caffeine administration was initiated on the 3rd day of life, but this had little effect on apnea. Apnea and cyanosis mostly occurred during bottle-feeding.
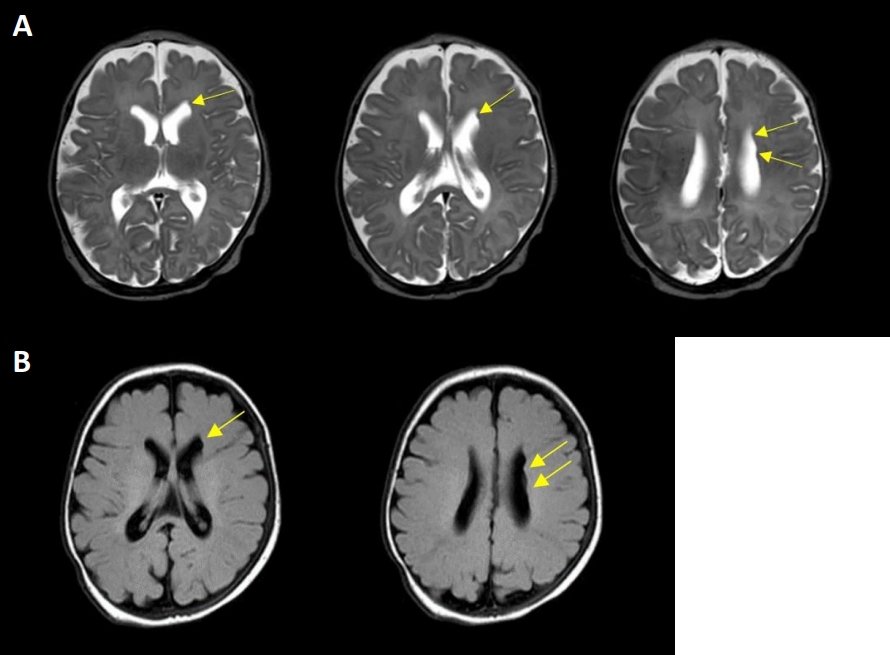
A comprehensive evaluation was performed to determine the cause of persistent feeding cyanosis and apnea. No abnormalities were observed in the blood tests, including complete blood count, serum chemistry, and electrolytes. Ultrasonography of the brain revealed no abnormalities. The patient showed no clinical seizures, and electroencephalography (EEG) was performed to identify whether apnea was a symptom of subclinical seizure; however, EEG showed no positive findings. A video fluoroscopic swallowing study (VFSS) was performed to determine whether feeding cyanosis was due to aspiration; however, there were no positive findings. Brain MRI was performed at 39+4 weeks of postmenstrual age, showing several subependymal nodular lesions with isosignal intensity of gray matter projecting into the left lateral ventricular wall (Figure 1). As FLNA is widely known to cause PNH, genetic studies have been performed using nextgeneration sequencing (NGS)-based gene panel tests. The gene panel included 652 genes associated with genetic epilepsy, including FLNA; however, there were no pathogenic or likely pathogenic variants of FLNA in this patient. Whole-genome sequencing of the patient and her parents was performed, although the results have not yet been confirmed.
At the postmenstrual age of 52+2 weeks, she was able to feed with a Special Needs Feeder from Medela Corp. (Baar, Swiss), and was discharged. At the time of discharge, her body weight was 5,265 g (24 percentile), body height was 55.8 cm (4 percentile), and head circumference was 39.3 cm (48 percentile). She did not show any signs of developmental delay, except for poor sucking. After discharge, the patient used a Special Needs Feeder from Medela Corp. for 1 month and was able to successfully change to a normal milk bottle. She now has a postnatal age of 9 months, does not have any problems with feeding, and has started eating baby food. The patient successfully achieved normal growth, with a body weight of 6,800 g (20 percentile), body height of 66 cm (30 percentile), and head circumference of 42.2 cm (25 percentile). She does not currently show any neurological symptoms or signs such as developmental delay or seizures. We are currently following up the patient every 3 months to check her neurological development and feeding status.
DISCUSSION
During early brain development, neuronal precursor cells migrate from their origin along the ventricles to the brain cortex. Failure of this process may cause nodular heterotopia (NH), and the most common form of NH is PNH, also known as subependymal NH [1].
The prevalence of PHN is unknown, but PNH has been reported in 11% to 20% of patients with cortical malformations and epilepsy [4]. PNH can be diagnosed radiologically using various imaging techniques, such as ultrasonography and computed tomography, but is most easily diagnosed using MRI [2]. Imaging alone can confirm the diagnosis of PNH but cannot identify specific genetic mutations. Genotyping or exonic sequencing for absolute identification of cause is required [3].
Patients with PNH show a wide spectrum of clinical findings, ranging from asymptomatic with normal intelligence to severe intractable epilepsy with developmental and intellectual disabilities [5]. Epilepsy was found in 72% of patients with PNH, with a mean age of 12 years [6]. PNH may also show some central nervous system anomalies, including corpus callosum agenesis, hydrocephalus, or lissencephaly, and cardiovascular abnormalities, such as aortic valve insufficiency or PDA [7]. This patient had symptomatic PDA that had to be ligated surgically; however, she did not show any signs of PNH during antenatal care.
The patient had recurrent apnea and longstanding cyanosis. There have been no reports of these symptoms in infants with PNH. There has been a report of PNH presenting with respiratory distress symptoms in a 2-month-old female infant; however, interstitial lung disease was found in this case, suggesting scimitar syndrome [8]. The EEG of the patient in the present study revealed that apnea and feeding cyanosis were not isolated manifestations of epileptic seizures, and VFSS confirmed that she did not have any motility problems.
The main differential diagnosis for PNH is tuberous sclerosis, which can involve subependymal tubers; however, these can be differentiated because lesions in tuberous sclerosis are often calcified, irregular in shape, and enhanced after gadolinium administration [9].
PNH can be found in isolated gene defects; however, it seems to be a genetically heterogeneous disease involving many different genes, such as FLNA, ARFGEF2, neural precursor cell expressed, developmentally downregulated 4-like (NEDD4L), and microtubule-associated protein 1 B (MAP1B) [7]. Additionally, PNH is associated with many syndromes, including Ehlers-Danlos, Fragile X, and Williams syndrome [1,7]. FLNA-related X-linked PNH shows rather specific bilateral heterotopia in combination with cerebellar hypoplasia and corpus callosum abnormalities [10]. ARFGEF2 mutations can cause bilateral heterotopia and severe neurologic deficit [7]. It is not surprising that our patient had a negative FLNA test result. Additionally, chromosomal abnormalities have been reported to be the cause of recurrent or single cases of PNH [7]. In a recent study, diagnostic work-up of patients with cortical development malformations found 212 involved genes using NGS, 16 of which were associated with PNH [11]. Another study reported a diagnostic yield of 36% when chromosomal microassay analysis was performed in patients with PNH [12]. Because symptoms of PNH are not usually apparent in infancy, it is difficult to predict which gene was involved in our case, and we are currently waiting for the results of whole genome sequencing of the patient and her parents. Although not fully understood, there may be nongenetic causes of PNH. Acquired insults in the developing brain, such as ischemia, may cause neuronal migration failure and eventually lead to NH [9].
In summary, we report the case of a newborn infant born at 34 weeks of gestation who had symptomatic PDA, feeding cyanosis, recurrent apneic spells, and unilateral PHN diagnosed by brain MRI. No mutations in FLNA were detected by NGS and no chromosomal abnormalities were identified. Whole-genome sequencing is currently in progress to clarify the patient’s genetic background. Cyanosis and apneic spells have not been previously reported in newborn infants with PNH. Thus, brain MRI as a diagnostic tool for identifying such brain abnormalities may be an option for newborns with prolonged apneic spells.