First Successful Application of Preimplantation Genetic Diagnosis for Lethal Neonatal Rigidity and Multifocal Seizure Syndrome in Korea: A Case Report
Article information

Abstract
Lethal neonatal rigidity and multifocal seizure syndrome (RMFSL) is a severe autosomal recessive epileptic encephalopathy characterized by rigidity, intractable multifocal seizures, microcephaly, apnea, and bradycardia immediately after birth. RMFSL is related to a mutation in breast cancer 1-associated ataxia telangiectasia mutated activation-1 protein (BRAT1). We report a case of a female infant born to non-consanguineous Korean parents who developed hypertonia, dysmorphic features, progressive encephalopathy with refractory seizures at birth, and worsening intermittent apnea, leading to intubation and death at 137 days of age. The initial repeated electroencephalographic findings were normal; however, a pattern of focal seizures emerged at 35 days of life. Rapid trio whole-exome sequencing revealed heterozygous mutations c.1313_1314delAG p.(Gln438Argfs*51) and c.1276C>T p. (Gln426*) in BRAT1. After genetic counseling for pregnancy planning, a preimplantation genetic diagnosis for targeted BRAT1 mutations was successfully performed, and a healthy baby was born. To our knowledge, this is the first reported case of a Korean patient with compound heterozygous mutations in BRAT1. An early and accurate genetic diagnosis can help provide timely treatment to patients and indicate the need for reproductive counseling for parents for family planning.
INTRODUCTION
Lethal neonatal rigidity and multifocal seizure syndrome (RMFSL, OMIM #614498) is a severe epileptic encephalopathy characterized by rigidity and multifocal intractable seizures, microcephaly, and apnea, as well as severe bradycardia that leads to cardiac arrest and death. This syndrome is caused by a mutation in the breast cancer 1-associated ataxia telangiectasia mutated activation-1 protein (BRAT1) gene [1,2]. There are a few reports of this syndrome in the literature, most of which were cases of infants born to consanguineous parents who died within a few months of their 1st year of life. Herein, we report the first Korean neonate who was diagnosed with compound heterozygous BRAT1 mutations and the successful application of preimplantation genetic diagnosis (PGD) for targeted BRAT1 mutations for the next pregnancy.
CASE REPORT
A female infant was born to non-consanguineous parents at 40 weeks plus 1 day of gestation. The findings of several intrauterine ultrasound examinations performed during pregnancy were normal. The patient was delivered via emergency cesarean section because of failure to progress in labor. The Apgar scores were 8 and 9 at 1 and 5 minutes, respectively. Her birth weight was 3,310 g (40th percentile, –0.26 standard deviation [SD]), her length was 49 cm (25th percentile, –0.69 SD), and her head circumference was 32.5 cm (5th percentile, –1.65 SD). Shortly after birth, the patient was noted to have frequent myoclonic seizure-like activity with hypertonia. Microcephaly with a small anterior fontanelle, clubfoot, low-set ears, and small forehead was observed at birth. The patient was subsequently transferred to a tertiary hospital for further evaluation. On admission to the neonatal intensive care unit, the patient was markedly hypertonic with contractions of the elbows and wrists and scissoring of both lower limbs. Neonatal reflexes were not elicited because of the increased muscle tone. Myoclonic jerks were prominent in the extremities and face in response to touch.
Electroencephalography (EEG) and brain magnetic resonance imaging (MRI) were performed on the 1st day of life. The EEG results were unremarkable. However, brain MRI revealed a diffusely decreased cerebral volume with a markedly prominent subarachnoid space and operculum widening with normal myelination. There were no abnormal signal changes in either the basal ganglia or the thalami (Figure 1A-D). Toxoplasmosis, rubella, cytomegalovirus, herpes simplex, and other septic workup were all negative, and findings on comprehensive neurometabolic tests such as neonatal screening test, amino acid, lactate, ammonia, acylcarnitine profile, urinary ketones, and quantitative amino acid and organic acids in urine did not reveal any abnormalities.
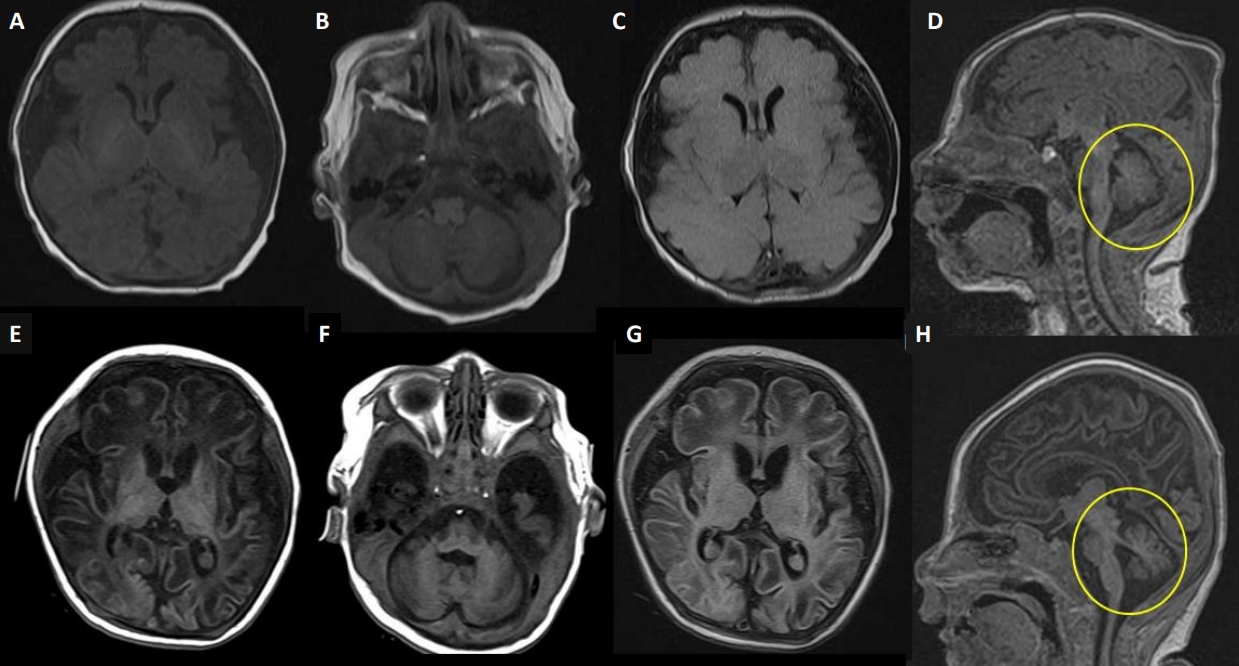
Brain magnetic resonance imaging (MRI) scans. Upper row at day 1 of life (A-D), lower row at 2 months (E, F). (A, B, E, F) Axial T1-weighted images; (C, G) axial T2-weighted axial fluid-attenuated inversion recovery (FLAIR) images; and (D, H) sagittal T1-weighted images. (A-D) Brain MRI on day 1 showed a diffusely decreased volume of the cerebrum with a markedly prominent subarachnoid space and operculum widening with normal myelination. However, there were no demonstrable abnormal signal changes in the basal ganglia or the thalami. (E-H) Brain MRI at 2 months showed suspicious cystic encephalomalacia changes in bilateral parietal areas and progressive atrophy of the cerebrum, cerebellum, and brainstem (circles on E, F).
Hypertonia with rigidity and myoclonic seizure-like activities worsened, and the patient was eventually intubated at 25 days of age because of frequent apnea, bradycardia, and desaturation. Phenobarbital and levetiracetam were inefficacious, baclofen and clonazepam were added to treat hyperekplexia. Serial routine EEGs revealed normal results during the 1st month of life. At 35 days of age, we observed frequent clinical seizures with ictal activity from video-EEG monitoring. Serial video-EEG follow-ups revealed multifocal medium-to-high-voltage sharp wave discharges, predominantly on the left of the right central regions (Figure 2). As the patient did not respond to antiepileptic drugs, including phenobarbital, levetiracetam, baclofen, clonazepam, valproate, and vitamin challenges, a continuous infusion of midazolam was administered at approximately 43 days of life. Although midazolam was partially effective, the seizures were not completely controlled. We performed chromosomal microarray and rapid trio whole-exome sequencing for decision making regarding early onset intractable myoclonic seizures, hypertonia, and microcephaly. Serial brain MRI at 65 days of age revealed suspicious cystic encephalomalacia changes in bilateral parietal areas and interval-progressed atrophic changes in the cerebellum and brainstem (Figure 1E-H).

Electroencephalography (EEG) findings. Ictal EEG at 35 days showing repetitive medium-to-high-voltage sharp wave discharges (arrows) from the right central region to the right hemisphere.
During the admission period, the patient’s weight and length remained within the normal range, while her head circumference decreased below the 3rd percentile (Figure 3). The patient had feeding problems necessitating a nasogastric tube owing to poor oral sucking and recurrent aspiration.

Growth curves of the patient. (A) Growth curves showing the patient’s body weight and length. (B) Growth curve showing the patient’s head circumference.
At 89 days of age, rapid trio whole-exome sequencing revealed a heterozygous mutation in the BRAT1 gene, c.1313_1314delAG p.(Gln438Argfs*51) and c.1276C>T p.(Gln426*) (NM_152743.4), which were subsequently validated by Sanger sequencing (Figure 4). This mutation has previously been reported in the literature and is regarded as pathogenic. The patient died 137 days after birth. An autopsy was not performed according to the parents’ requests. For subsequent planning of pregnancy, we recommended PGD for the BRAT1 gene. Only unaffected embryos were transferred, and singleton pregnancy was achieved. At 12 weeks, chorionic villus sampling was performed for re-confirmation of the genotype of transferred embryos, and an unaffected fetus was confirmed by the polymerase chain reaction method. Consequently, a healthy girl was born.

Pedigree and Sanger sequencing. Pedigree of the family. Wild-type alleles are indicated using +. A compound heterozygous mutation in breast cancer 1-associated ataxia telangiectasia mutated activation-1 protein (BRAT1) validated by Sanger sequencing. The frameshift mutation (c.1313_1314delAG) was found in the father and the patient, and the nonsense mutation (c.1276C> T) was confirmed in the mother and the patient.
DISCUSSION
RMFSL is a severe epileptic encephalopathy caused by BRAT1 gene mutations [1]. BRAT1 encodes a protein that interacts with tumor suppressor breast cancer gene 1 (BRCA1) and binds to ataxia telangiectasia mutated 1 gene (ATM-1) [2]. It is involved in cell cycle signaling pathways required for cellular responses to DNA damage [2]. The neuropathological examination has been reported in five cases, showing progressive atrophy and neuronal cell loss in the cerebrum and cerebellum, suggesting that disturbed apoptosis may occur because of BRAT1 mutations [3,4]. Several recent studies have demonstrated that loss of BRAT1 inhibits growth signaling, increases apoptosis, and induces mitochondrial dysfunction, which is involved in mitochondria-dependent intrinsic apoptosis [5].
The clinical features of RMFSL include progressive encephalopathy with hypertonia and hyperreflexia, microcephaly, various dysmorphic features, and developmental delay. After Puffenberger et al. [1] first reported three Amish infant siblings homozygous for the BRAT1 frameshift mutation (c.638_639insA) in 2012, an additional 24 severe cases of the spectrum (a total of 27) have been reported. Patients with the severe type are described and compared to our case in Table 1 [1,3,4,6-18]. A severe phenotype of RMFSL characterized by prenatal or neonatal onset of refractory myoclonic seizures, episodes of apnea, and absence of developmental progression leads to death within weeks to months of birth [3]. All EEGs were previously reported to be abnormal at the time of the evaluation. EEG typically shows diffuse slowing, consistent with encephalopathy, and (multi)focal sharp wave activity, consistent with ictal activity. Most brain MRI results were normal at the initial evaluation and demonstrated variable degrees of cerebral and/or cerebellar atrophy in serial follow-ups [4,14,15,17]. In our case, the clinical presentation was similar to that of other severe phenotypes. MRI findings showed significant cerebral atrophy on the 1st day of life, potentially indicating the sequelae of chronic exposure to refractory seizures during the prenatal period. However, serial routine EEGs during the 1st month of life did not reveal any abnormalities. Since the EEG findings were not pathognomonic during the 1st month and there were no other structural, metabolic, or infectious causes, we initially considered the symptoms presented in this case to represent hyperekplexia. However, the patient’s hypertonia with rigidity and refractory myoclonic seizures worsened, and invasive ventilator support was required for frequent apnea, bradycardia, and desaturation. Epilepsy of a genetic origin, such as RMFSL, was suspected. Because BRAT1 was not included in the neonatal-onset genetic epilepsy panel, we performed rapid trio whole-exome sequencing for decision-making at 35 days of life, and the results were obtained at 54 days.

Characteristics of 27 Previously Reported Cases with Severe Clinical Form of BRAT1-Related Rigidity and Multifocal Seizure Syndrome
The importance of genetic testing has recently been emphasized. New genes responsible for neonatal-onset epilepsy have been rapidly discovered, and BRAT1 has been shown to cause RMFSL [16,17]. Shellhaas et al. [19] reported that more than three-quarters of the neonates with epileptic encephalopathies were genetically confirmed. Because many neonatal epilepsy syndromes have a genetic etiology, genetic testing should be considered as soon as possible to provide targeted treatments [19]. In addition, for the next pregnancy, genetic counseling of the parents should be considered. PGD can provide an alternative to prenatal diagnosis and selective abortion for parents at risk of transmitting a serious genetic disorder. Oocytes or cleaving embryos obtained from in vitro fertilization and biopsied cells were used for genetic diagnosis. Only embryos identified as free of genetic disorders were transferred into the uterus [20].
In our case, the patient presented with a severe RMFSL phenotype and was determined to have a heterozygous nonsense or frameshift mutation in the BRAT1 gene. PGD was successfully applied to the next pregnancy, and a healthy baby was born. To our knowledge, this is the first reported case of successful PGD for BRAT1 mutations in Korea.
In conclusion, genetic studies can be helpful for patients with severe epileptic encephalopathy. This report highlights the role of rapid trio whole-exome sequencing in early diagnosis and genetic counseling. Early and accurate genetic diagnosis can provide timely management to patients and indicate the need for reproductive counseling for parents for planning the next baby.
Notes
Ethical statement
This study was approved by the Institutional Review Board of Seoul National University Bundang Hospital (IRB approval number: B-2206-763-701), and written informed consent was obtained from the patient’s parents. The patient’ parents provided informed consents for the publication of the present report.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conception or design: G.E.Y., Y.H.J.
Acquisition, analysis, or interpretation of data: G.E.Y., Y.H.J.
Drafting the work or revising: G.E.Y, Y.H.J., S.Y.K., S.A.C., H.K., C.W.C.
Final approval of the manuscript: All authors read and approved the final manuscript.
Funding
None
Acknowledgements
None