INTRODUCTION
Schaaf-Yang syndrome (SYS), which shares phenotypic features with Prader-Willi syndrome (PWS), is a rare genetic disorder caused by loss-of-function variants of MAGE family member L2 (MAGEL2) in the paternally derived allele. Since its first descrip┬Łtion in 2013 [1], more than 250 patients have been reported in the literature [2]. Neonatal hypotonia, developmental delay, intellectual disability, and short stature are common characteristics of both SYS and PWS. However, the phenotype of SYS differs from that of PWS in that the prevalence of respiratory distress in early infancy, distal joint contractures, and autistic features are more common in SYS, whereas the hyperphagic phase is less common [3]. Genetic diagnosis of SYS is established when a heterozygous loss-of-function variant of MAGEL2 in the paternal allele is identified in a proband, as MAGEL2 is a maternally imprinted gene [4]. Here, we describe a case of SYS that presented with respiratory distress at birth, micro┬Łpenis, cryptorchidism, transient renal dysfunction, hearing impairment, and growth and developmental retardation.
CASE REPORT
A boy aged 3 weeks was transferred to our neonatal inten┬Łsive care unit (NICU) for evaluation of airway problems to determine whether tracheostomy was necessary. After birth, the patient was admitted to the NICU because of respiratory distress. Non-invasive positive pressure ventilation was initial┬Łly applied, followed by intubation and mechanical ventilation, 7 hours after birth. Thereafter, the extubation trial failed recurrently even though the ventilator setting was minimal. At 12 days of age, hypoplasia and deflection of the epiglottis were found on bedside laryngoscopy; however, there was no significant subglottic stenosis. The patient was the first boy from a single pregnancy born to a non-consanguineous couple. The pregnancy was uneventful, except that poly┬Łhydramnios was noted for the first time, 8 days prior to delivery. The baby was delivered at a gestational age of 38 weeks and 6 days, with a birth weight of 3,330 g.
On admission, micrognathia, retrognathia, cleft palate, arthrogryposis of both hands, micropenis, and an undescend┬Łed right testis were observed. Direct laryngoscopy with bronchoscopy conducted at the age of 24 days showed de┬Łflection of the epiglottis and absence of glottic or subglottic stenoses. Tracheomalacia and bronchomalacia of both main bronchi were noted during the examination. Therefore, tracheostomy was immediately per┬Łformed to bypass signifi┬Łcant airway obstruction at the level of the deflected epiglottis. After the surgery, full oral feeding was achieved. No structural anomalies were found on the sonographic evaluation of the brain, heart, or renal system. There were no abnormal findings on ophthalmological examination. The automated auditory brainstem response test did not reveal clear res┬Łponses in either ear. He was discharged at 43 days of age with O 2 supplementation at 0.05 L/min via a tracheostomy tube.
Chromosomal analysis revealed a heteromorphic chromosome 9 (46/XY, 9qh+), and no duplication or deletion was detected in the chromosomal microarray. At 4 months of age, an auditory brainstem response threshold (ABRT) test was performed, and the results of the right and left ABRT were 45 and 40 dB, respectively, compatible with moderate hearing loss (41 to 55 dB). Tympanometry showed normal middle ear status, and middle ear effusion was absent in both ears. At 6 months of age, his body length was 60 cm (<1st percentile), weight was 6.8 kg (8th percentile), and head circumference was 43.2 cm (45th percentile). Skeletal dysplastic features, such as a narrow thorax, tapered fingers, finger contractures, and short toes, became more prominent. He was able to control his head at 6 months of age and roll from front to back at 9 months of age. However, he could not roll from back to front or crawl on his belly at 10 months of age. Eye contact and two-hand transfer was not noticeable until 10 months of age.
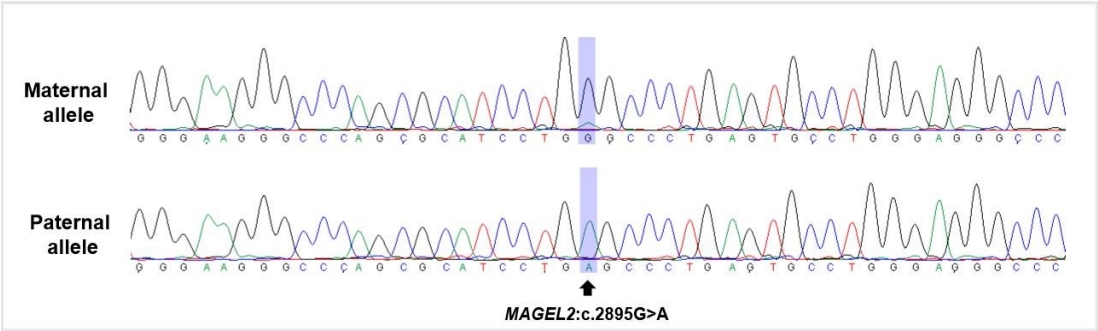
To rule out hereditary skeletal disorders, gene panel tests encompassing 31 causative genes for spondyloepiphyseal and metaphyseal dysplasia were conducted, which revealed no meaningful variants. Whole-exome sequencing (WES) of the patient revealed a previously reported heterozygous nonsense variant, NM_019066.5(MAGEL2):c.2895G>A (p.Trp965*), which was confirmed as a de novo variant using additional Sanger sequencing of MAGEL2 gene in both parents. To confirm the diagnosis, allele-specific polymerase chain reaction (PCR) and sequencing were performed using single nucleotide poly┬Łmorphism rs9785. The variant was revealed on the paternal allele of MAGEL2 and the patient was confirmed to have SYS (Figure 1).
Decreased renal function (estimated glomerular filtration rate [eGFR] of 52 mL/min/1.73 m2 using the Schwartz equation) was first noted at the age of 10 months and re┬Łmained stationary thereafter (mild renal dysfunction: eGFR <60 mL/min/1.73 m2). The patient did not experience a definite episode of acute kidney injury. Although congenital anomalies of the kidney and urinary tract were not demon┬Łstrated in repeated evaluations, the length of both kidneys was relatively small compared with age-matched normal populations at the age of 10 months (right 5.25 cm, left 5.23 cm; normal range, 5.32 to 6.96 cm). As micropenis and right cryptorchidism were still noted until 10 months of age, the human chorionic gonadotropin (hCG) stimulation test was conducted which showed an incremental response of testosterone indicative of normal testicular function (0.18 ng/mL at baseline and 2.42 ng/mL after stimulation). Orchiopexy has not yet been performed. At 19 months of age, the patientŌĆÖs body length was 63.3 cm (<1st percentile) and weight was 7.1 kg (<1st percentile). The growth velocity was 3.1 cm/year, which was markedly lower than that of age- and sex-matched healthy controls. In the Bayley Scales of Infant DevelopmentŌĆö3rd edition, at 19 months of age, developmental delay was found in the cognitive, language, and motor domains (raw scores of 26, 12, and 45; compatible with the developmental ages of 6, 3, and 6 months, respec┬Łtively). At the latest follow-up (aged 24 months), the tracheo┬Łstomy cannula was still present, and the patient underwent physical rehabilitation. He regularly visited our outpatient clinic to address and resolve respiratory, developmental delay, hearing, and growth retar┬Łdation issues. Palatoplasty is planned to help the patient articulate.
DISCUSSION
The present study describes the case of a Korean boy dia┬Łgnos┬Łed with SYS, harboring the truncating variant p.Trp965* in MAGEL2. To date, more than 250 individuals have been reported as being diagnosed with SYS [2]. By May 2022, 57 molecular variants within the MAGEL2 gene were classified as pathogenic or likely pathogenic according to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar). The c.1996dup (p.Q666fs) variant of MAGEL2 is the most common causative variant found in approximately half of known individuals with SYS [2]. In Korea, four cases of SYS have been reported, with two patients harboring the variant c.1996dup [5]. In the present report, a heterozygous truncating variant of c.2895G>A (p.Trp965*) in MAGEL2 was detected by WES, which was found to be a de novo variant by Sanger sequencing of samples from both parents. Since MAGEL2 is a maternally imprinted gene, deter┬Łmining the origin of the pathogenic variant was required for diagnosis. Allele-specific PCR demonstrated that the variant was paternally derived in the present case, confirming the diagnosis of SYS. The variant c.2895G>A found in our patient was a variant previously reported by Ravenscroft et al. [6], which resulted in an identical protein change (p.Trp965*) as another variant of c.2894G>A [7,8]. However, the parental origin of this gene has not been evaluated in previous reports. As with other imprinting disorders, the origin of the genetic variant should be documented to diagnose SYS in an index case with a de novo variant because variants in the maternal MAGEL2 allele are clinically insignificant. Although a heteromorphic variant of chromosome 9 was identified in the present case, it is considered a normal variant, with the reported prevalence being in the range of 1% to 2% in the general population [9,10]. Only a few studies have reported that it is accompanied by idiopathic reproductive failure in female patients [11]. There┬Łfore, the association between heteromorphic chromo┬Łsome 9 and clinical manifestations in the present case may not be valid.
Our patient required intubation on the first day after birth because of significant respiratory distress. The cause of re┬Łspiratory distress may be explained by structural problems in the upper airway. After performing tracheostomy, the patient did not show any respiratory difficulties after weaning from pressure support via a ventilator. In the largest retrospec┬Łtive cohort study of 78 patients with SYS, 18% of patients required tracheostomy [3]. In 2018, Matuszewska et al. [7] reported a case of a female patient with SYS with c.2894G>A, who was treated with therapeutic hypothermia for perinatal hypoxic events immediately after birth and underwent tracheostomy for respiratory failure associated with generalized hypotonia. Distal arthrogryposis was observed on the day of birth. In 2020, Smigiel et al. [8] reported another female patient with SYS with c.2894G>A, who had signs of respiratory failure, arthrogryposis, and hypotonia at birth. In 2021, Ravenscroft et al. [6] reported a patient harboring c.2895G>A in MAGEL2 who had distal arthrogryposis, obstructive sleep apnea, and feeding difficulties; however, the onset of symptoms was not described in detail. Our patient and two previously reported patients with c.2894G>A showed clinical signs of respi┬Łratory difficulty, arthrogryposis, and hypotonia on the first day of birth (Table 1).
A significant proportion of patients with SYS experience hypo gonadism, although it is not as common as that in PWS [2,12,13]. For patients with SYS, there is a lack of evidence regarding the use of hormone replacement therapy (HRT) to treat micropenis and cryptorchidism or to induce puberty. In the present study, the patient had micropenis and cry┬Łptorchidism, and the hCG stimulation test was performed to evaluate the ability of Leydig cells to produce androgens, which showed a normal incremental response. Based on the positive hCG stimulation test results and an increase in phallus length during serial examinations, the implementation of HRT was waived.
Our patient showed mild renal dysfunction (eGFR <60 mL/min/1.73 m2) accompanied by small size of both kidneys, which was first noted at 10 months of age and did not im┬Łprove or deteriorate thereafter. There were no episodes of acute kidney injury during the neonatal period. The relationship between decreased renal function and genetic problems is unclear, given that MAGEL2 is highly expressed restrictively in the human brain but not in the kidneys [4,14]. Therefore, a long-term follow-up of the present case is required. Hearing impairment was also found in our patient at 4 months of age without any evidence of middle ear pro┬Łblems. Currently, little is known about the incidence and causes of hearing problems in patients with SYS. As renal and hearing manifestations of SYS have not been reported thus far, further follow-up of the patient is warranted.
Here, we describe a patient with SYS harboring a de novo variant, MAGEL2:c.2895G>A (p.Trp965*), in the paternally derived allele, which is the first report of such a finding in the Korean population. We suggest that the variant p.Trp965* of MAGEL2 may be associated with the clinical manifestation of hearing impairment in SYS.