INTRODUCTION
Cleidocranial dysplasia (CCD) is a rare autosomal dominant skeletal dysplastic disease first described by Marie and Sainton [1] in 1898, with an incidence rate of 1:1,000,000 [2]. CCD is associated with mutations in the gene that encodes the Runt-related transcription factor 2 (RUNX2), which is essential for the differentiation of periosteal cells into osteoblasts [3]. CCD is characterized by hypoplastic clavicles, patent cranial sutures and fontanelles, supernumerary teeth, and facial features such as frontal bossing and hypertelorism [4]. The presence of such features on prenatal abdominal ultrasound is suggestive of CCD [5]. After birth, palpation of the skull may reveal wide, soft portions where fontanelle closure has not occurred. There is no specific treatment for CCD; however, management is essential to prevent complications (such as blunt trauma to the brain, upper airway obstruction, and injury to the brachial plexus and vessels) during growth of the child. In the long term, orthodontic or surgical treatments such as removal of supernumerary teeth may be required [4]. In this study, we have reported the case of an infant diagnosed with CCD after birth that was followed up for 4 years.
CASE REPORT
A male infant was delivered by elective cesarean section at a gestational age of 37 weeks and 5 days. His motherŌĆÖs parity was one preterm delivery, two artificial abortions, and one live birth. His brother was born prematurely due to maternal pre-eclampsia, and there were no deformities. Prenatal examination during the current pregnancy showed intrauterine growth retardation and a short femur than that expected for the gestational age. However, there were no abnormal findings in other bones; therefore, the infant was admitted to the neonatal intensive care unit (NICU) following delivery for evaluation. His Apgar score was 7 at 1 minute after birth and 9 at 5 minutes. His birth weight was 2,730g (between the 25th and 50th percentiles), body length was 45 cm (between the 10th and 25th percentiles), and head circumference was 33.5 cm (25th percentile). The infant did not require resuscitation in the delivery room; however, shortness of breath, cyanosis, and chest retraction were observed after admission to the NICU. Endotracheal intubation was performed and ventilator care was provided.
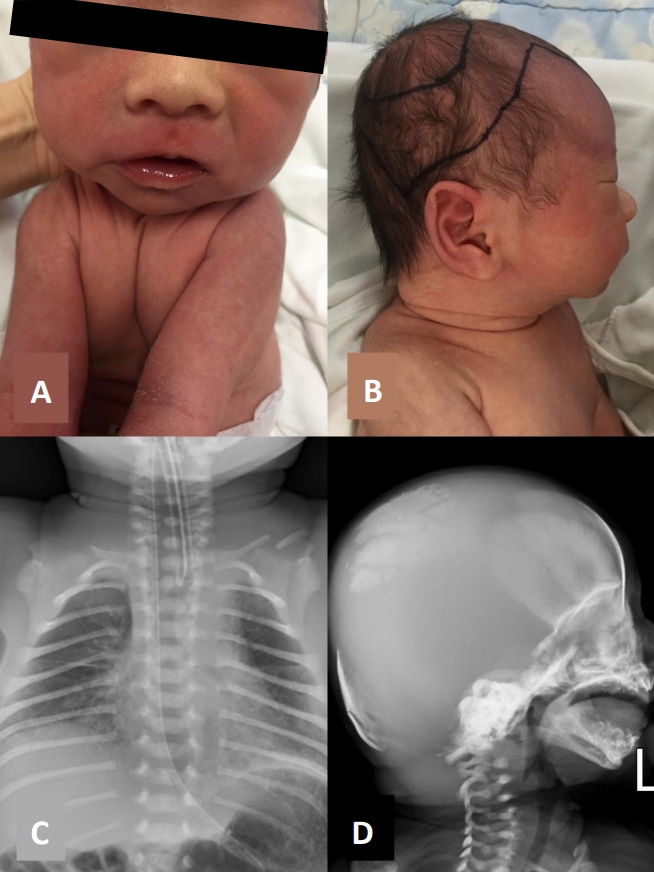
Physical examination revealed unusually large fontanelles, with wide suture lines and hypertelorism (Figure 1A, B). It was difficult to distinguish between the clavicles on both sides during palpation. The shoulders were narrow, and could be moved closer to each other compared to other normal infants (Figure 1A). Laboratory examination revealed normal levels of total calcium and phosphorus.
A chest radiograph showed incomplete expansion of the lungs with bilateral ground-glass opacity, as well as hypoplasia of both clavicles (Figure 1C). A skull radiograph showed hypoplasia of the frontal, occipital, parietal, and temporal bones (Figure 1D). An infantogram was taken to confirm the presence or absence of other bones, which did not reveal any abnormalities in the length and shape. The length of the femur was 67 mm (reference, 66 to 76), which was within the normal range.
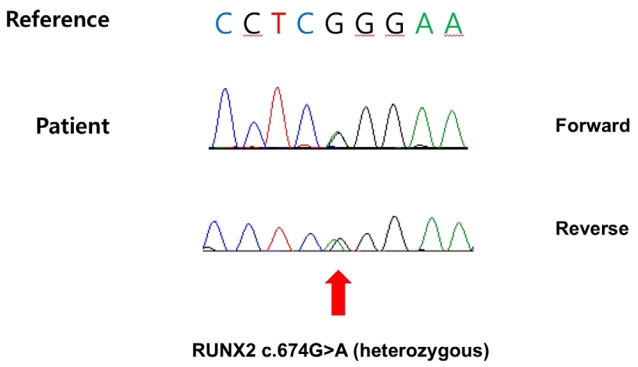
These clinical findings were considered likely signs of CCD, and a mutational analysis of the RUNX2 gene was conducted to confirm the diagnosis, with consent from the parents. The analysis was performed by direct sequencing of eight exons of the RUNX2 gene. A heterozygous mutation was observed in the Runt domain c.674G>A (p.Arg225Gln), which was previously identified as a pathogenic variant [6], thereby confirming the diagnosis of CCD (Figure 2). The parents did not have any family history of genetic disorders or clinical features of CCD. The motherŌĆÖs chest radiograph did not reveal any bone abnormality. The parents refused to undergo genetic testing; therefore, hereditary characteristics could not be confirmed.
On admission to the NICU, the infant was diagnosed with respiratory distress syndrome and was treated with surfactant therapy. By the 4th day after birth, his respiration had improved sufficiently for ventilator support to be discontinued. On day 7, vitals were evaluated and determined to be stable, following which the infant was discharged. Further follow-up was done in the outpatient department.
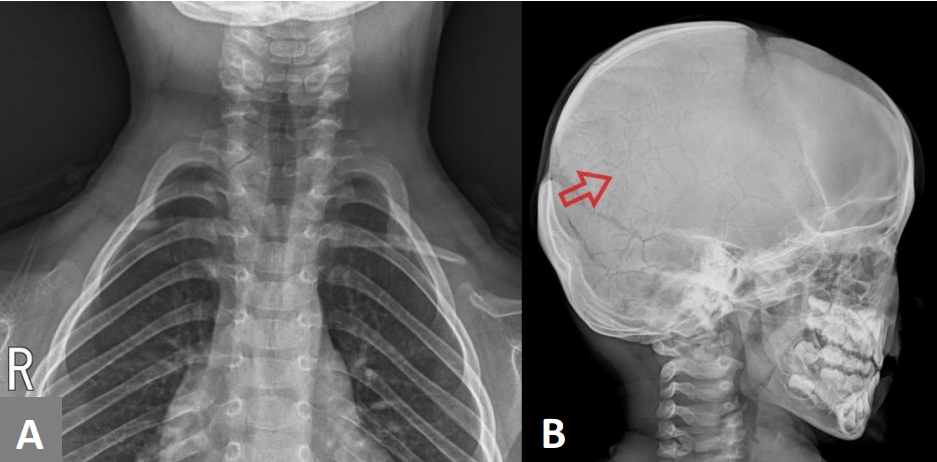
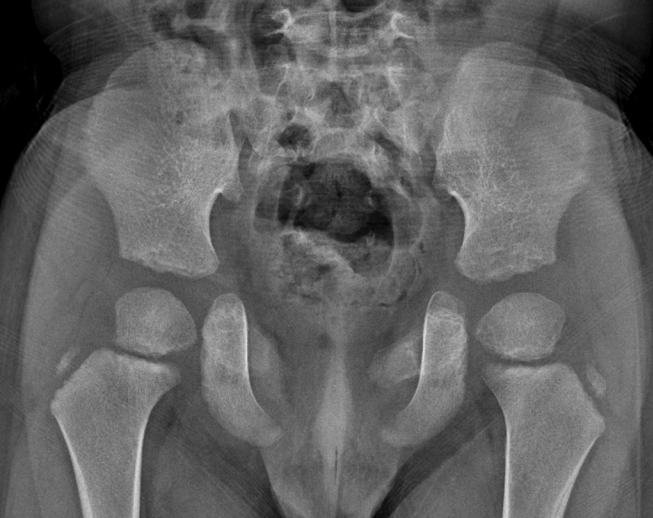
At 1 year of age, height (74.7 cm, 25 percentile), weight (9.6 kg, 25 to 50 percentile), and head circumference (46.5 cm, 50 to 75 percentile) of the child were within the normal range. Likewise, at 4 years of age, height (101.5 cm, 25 to 50 percentile), weight (17.4 kg, 50 percentile), and head circumference (51.6 cm, 50 to 75 percentile) of the child were within the growth curve. The bilateral dysplastic clavicles observed after birth remained underdeveloped (Figure 3A). A series of follow-up evaluations of the cranial bones during growth of the child revealed slower than usual skull development. However, most of the hypoplastic areas in the skull were covered with bones by 4 years of age. The cranial suture lines became narrower and multiple Wormian bones (intrasutural bones) appeared (Figure 3B). A wide symphysis pubis was also observed (Figure 4), which consistently remained wider than the mean for the childŌĆÖs age. However, there were no gait disturbances. Dental examinations were also performed annually from 6 months of age, wherein panoramic radiographs were taken. The teeth erupted normally according to age, and there were no supernumerary teeth or maxillary lesions.
The child was able to walk on his own after the age of 1 year. The Korean Developmental Screening Test for infants and children was performed at 2 years of age. Follow-up evaluations were recommended to assess the gross and fine motor abilities, cognition, communication, and social interaction skills. The childŌĆÖs self-control was within the peer level range. The Korean-Wechsler Preschool and Primary Scale of Intelligence-Fourth edition was used to assess intelligence when the child was 4 years old. His full-scale intelligence quotient was 91 points (reference, 90 to 109), which indicated average intelligence. Brain magnetic resonance imaging did not reveal any abnormality, and the child did not show any significant delay in development, including upper limb motor function.
Headgear was advised for protection as the child grew. The hypoplastic clavicle did not cause complications such as nerve or blood vessel injury, pain, or limitation of the range of motion. The childŌĆÖs chest was bell-shaped; however, he did not experience any respiratory symptoms due to the shape of the rib cage.
DISCUSSION
CCD is associated with characteristic clinical features; however, the degree of presentation varies in each patient. Final diagnosis of the condition is based on genetic testing for identification of mutations in the RUNX2 gene. The incidence of dental problems in patients with CCD is approximately 93.5% [7], and several domestic case studies have reported on the treatment of such problems. In contrast, in the current case, there were no complications other than skull and clavicle hypoplasia.
RUNX2 is a multidomain protein that regulates osteoblast differentiation through multiple signaling pathways [8]. Most RUNX2 mutations occur in the Runt domain, with missense mutations being the most common [9]. Although a study reported that mutations in the Runt domain were associated with higher chances of supernumerary teeth and maxillary hypoplasia [10], the phenotype-genotype correlation was unclear. The effects appear to be due to RUNX2 as well as other genes that interact with it [9]. However, in general, children with CCD have short stature and develop normal intelligence [2].
The features of CCD are detectable on prenatal ultrasound examination [11]. A positive family history has been reported in most recorded cases of prenatally diagnosed CCD [5,11,12]. Nonetheless, in this case, there was no family history to predict hypoplastic bone disease, and the only unusual finding on prenatal examination was shorter length of the femur than that expected for the gestational age. However, an unusually short femur is not known to be associated with CCD. Therefore, we recommend careful assessment of both clavicle shape and skull ossification in cases of family history of CCD or presence of a short femur in the fetus.
CCD can affect the entire skeleton; however, the skull, clavicle, and teeth are chiefly affected [4]. Although not seen in this case, dental complications such as supernumerary teeth are common and may require orthodontic or surgical treatment. Furthermore, a bell-shaped chest may be observed and dyspnea can occur due to decreased respiratory capacity [13]. Other features may include a short femoral neck and a wide symphysis pubis, which could lead to coxa vara and phalangeal abnormalities [14,15]. Hypoplasia of the skull and bilateral clavicles, a bell-shaped thoracic cage, and a wide symphysis pubis were observed in our patient.
Fontanelles usually close in early childhood; however, remain open throughout life in patients with CCD. Use of protective headgear is advised to such patients to prevent brain injury until the cranial sutures become narrow, which was seen in our patient at around 4 years of age. Clavicle removal surgery should be considered if the fragments cause symptoms such as pain or numbness [16]. Patients should be monitored continuously for respiratory symptoms and should undergo periodic checkup of skeletal development to determine the need for surgical management. Considering that CCD is an autosomal dominant disease, genetic counseling of parents is important to help them with family planning.
In conclusion, we have reported the case of an infant with CCD, who was diagnosed promptly after birth. The child underwent long-term medical monitoring in an outpatient setting until formation of the majority of the cranial bones, thereby preventing occurrence of complications.