INTRODUCTION
Neonatal diabetes mellitus (NDM) is a rare form of monogenic diabetes that is diagnosed within the first 6 months of life. It occurs in approximately 1:90,000 to 160,000 births, with at least 20 causative genes [1]. NDM can be transient or permanent. Transient neonatal diabetes mellitus (TNDM), which accounts for approximately half of the total NDM cases, usually resolves within 3 months of insulin therapy; however, it may recur as permanent diabetes later in life. Conversely, permanent neonatal diabetes mellitus (PNDM) requires lifelong insulin therapy [2]. TNDM is frequently caused by the overexpression of PLAGL1 and HYMA1 due to defects in 6q24, and the overall prevalence and presenting features vary with race and ethnicity [3]. More than 40% of PNDM cases are caused by mutations in either KCNJ11 or ABCC8 [4].
Continuous subcutaneous insulin infusion (CSII) and real-time continuous glucose monitoring (CGM) are advanced technologies that could be applied to any young patients with diabetes. Currently, CSII is the treatment of choice for the initial management of NDM because it allows the delivery of very small, accurate doses of insulin [1]. The utilization of a CGM device together with CSII reduces the need for frequent blood puncture and provides a minimally invasive method of 24-hour glucose monitoring.
Early genetic diagnosis and early management of NDM are crucial in understanding the clinical course and long-term outcome, as they affect the therapeutic decisions. However, most of the mutations causing NDM are rare recessive, which hinders their identification, and the genetic diagnosis of approximately 20% of NDM cases is still unknown [1]. Herein, we present a case involving an infant with overt progression of TNDM during the early postnatal period, managed with CSII and CGM.
CASE REPORT
A male infant, the only child of a nonconsanguineous Korean couple, was vaginally delivered at 38 weeks of gestation at a local hospital, with a birth weight of 2.7 kg (3rd to 10th centile) and a length of 49.0 cm (10th to 50th centile) [5]. Dysmorphic features were not observed. No family history of medical conditions, including diabetes mellitus, was declared. On day 2, he was referred to the neonatal intensive care unit (NICU) because of tachypnea (breath rate, 70/min) and hyperglycemia (blood glucose level, 381 mg/dL). His systolic and diastolic blood pressures were 74 and 42 mm Hg, respectively. His heart rate was 112 beats/min, and his body temperature was 36.4℃. His respiration rate normalized within 48 hours of admission to the NICU; however, he remained hyperglycemic (blood glucose level, 290 to 390 mg/dL). His serum C-peptide and insulin levels were 0.06 ng/mL and undetectable (<0.20 μU/mL), respectively (Table 1). His baseline chemistry, including serum blood urea nitrogen (15.3 mg/dL; normal, 7.0 to 20.0), creatinine (0.9 mg/dL; normal, 0.6 to 1.2), aspartate aminotransferase (38 U/L; normal, 14 to 40), and alanine aminotransferase (16 U/L; normal, 9 to 45), and complete blood count profiles were within the normal ranges. Urinalysis revealed a 4+ glucose level, but no ketones. Moreover, no sign of ketoacidosis was noted. The patient was screened for pancreatic autoantibodies (antibodies against glutamic acid decarboxylase, islet cell, islet antigen-2, and insulin) to rule out the possibility of type 1 diabetes, and tested negative. Liver and pancreas ultrasonography revealed no structural abnormalities.
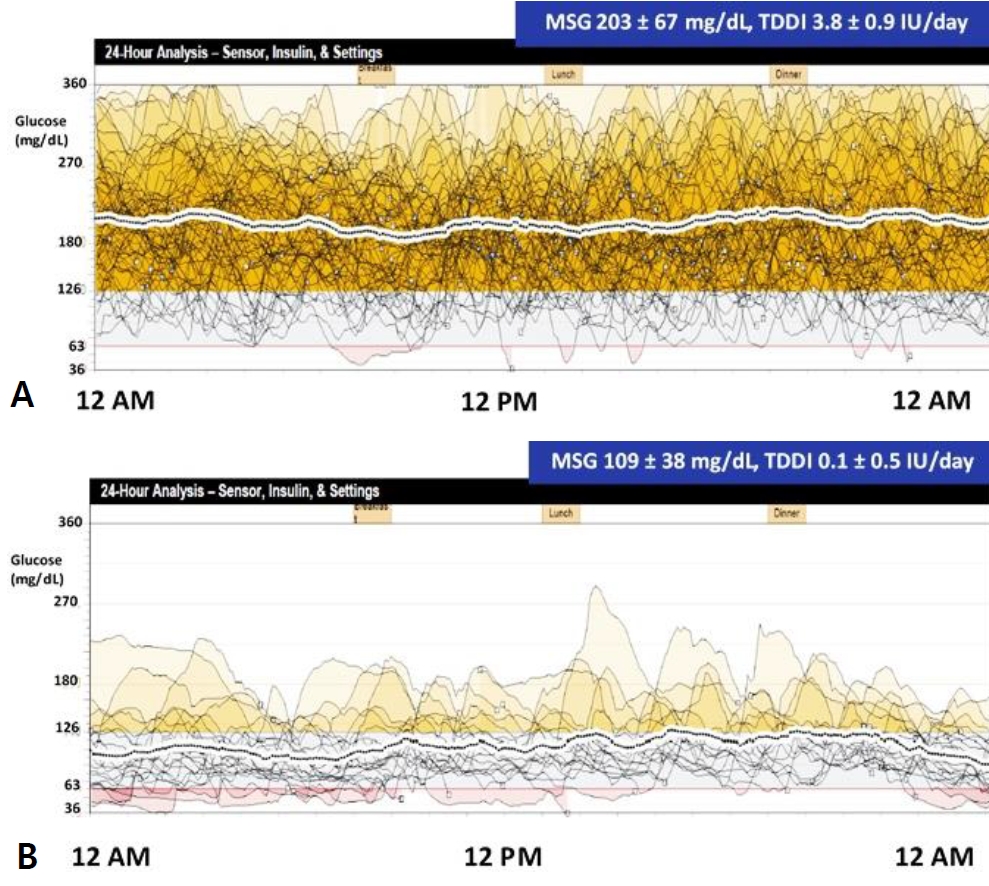
The initial clinical management of the patient was continuous intravenous administration of short-acting insulin (1.5 IU/day; Humulin R, Lilly Pharmaceutical, Indianapolis, IN, USA). The insulin infusion rate was transiently increased up to 3.9 IU/day for 0.5 hour at the start of every breastfeeding session. To monitor the glucose level and prevent hypoglycemia, fixed feeding intervals of 2.5 to 3.0 hours were recommended and blood glucose was measured using heel prick blood sampling at the commencement of each feeding session. Ultimately, 2.4 IU/day of insulin was required to maintain the blood glucose level in the range of 240 to 300 mg/dL. While awaiting the results of genetic testing, transition to oral sulfonylurea (SU) (0.2 mg/kg/day with gradual daily increases to 1.0 mg/kg/day while the insulin dose was decreased by 50% per day) was attempted to minimize potential neurodevelopmental disabilities associated with mutated ATP-sensitive potassium (K-ATP) channels [1]. The patient reverted to a hyperglycemic state. The SU trial treatment was discontinued 6 days after initiation, and the patient was switched back to intravenous insulin therapy, which resulted in a gradual improvement of the glycemic outcome. To facilitate continuation of treatment upon discharge, the transition from intravenous to subcutaneous insulin (MiniMed 640G, Medtronic, Minneapolis, MN, USA) administration was made 37 days postpartum. The pump was set to deliver a basal insulin amount of 3.0 IU/day, with an additional 0.2 to 0.5 IU of prefeeding bolus (preset square for 0.5 hour), active insulin time of 2.0 hours, predictive low-glucose suspend at 80 mg/mL, and high glucose alert at 400 mg/dL. The capabilities of the real-time CGM system in the latest model of the CSII pump are useful in eliminating heel pricks and episodes of hyperglycemia and hypoglycemia. The therapy was well tolerated, and the patient was discharged with the real-time CGM device and CSII pump on postnatal day 40 with an improved glycemic state and improved levels of C-peptide and insulin (Table 1, Figure 1). Upon discharge, his body weight was 5.0 kg (75th centile) and his length was 56 cm (75th to 90th centile) [5]. The mean sensor glucose (MSG), the direct interstitial fluid glucose level measured by the CGM device, was 203±67 mg/dL until the age of 3 months, with a total daily insulin requirement of 3.8±0.9 IU/day (Figure 2A). Subsequently, insulin therapy was no longer required and discontinued after 107 days. At the 4-month follow-up, the patient’s body weight was 7.6 kg (90th to 95th centile) and his length was 66 cm (90th centile). The patient reached normal 4-month developmental milestones (laughing aloud and loss of head lag), with no observed neurodevelopmental impairment. His glycemic status was more stable with an MSG level of 109±38 mg/dL (Figure 2B). Ultrasonography of the liver and pancreas was repeated, which showed no abnormal findings. In addition, the patient experienced neither skin troubles associated with subcutaneous devices nor hyperglycemic or hypoglycemic episodes, and his glycated hemoglobin level was 5.7% at age 6 months.
Genetic analysis was performed by isolating genomic DNA using a DNA sample preparation kit (TruSeq, Illumina, San Diego, CA, USA) and amplifying the enriched fragments of the target regions in the sequencing system (HiSeq2000, Illumina). The detection of insertions and deletions was based on an assigned algorithm (GATK RealignerTargetCreator, Broad Institute, Cambridge, MA, USA), and base scores were recalibrated (GATK BaseRecalibrator, Broad Institute). A targeted panel (ABCC8, BLK, CEL, eukaryotic translation initiation factor 2-alpha kinase 3 [EIF2AK3], FOXP3, GATA4, GATA6, GCK, HNF1A, HNF4A, HNF1B, INS, KCNJ11, KLF11, NEUROD1, PAX4, PDX1, pancreas-associated transcription factor 1-alpha [PTF1A], and ZFP57) sequencing for congenital diabetes identified two novel variants, as follows: a heterozygous missense variant (p.Arg1091Met, c.3272G>T) in exon 17 of the EIF2AK3 gene and a heterozygous missense variant (p.Ser18Phe, c.53C>T) in exon 1 of the PTF1A gene, both of which are classified as variants of uncertain significance (VUS) according to the American College of Medical Genetics guidelines [6]. Sanger sequencing confirmed the presence of both variants. The patient’s father was identified as a heterozygous carrier of the EIF2AK3 variant, whereas his mother was identified as a heterozygous carrier of the PTF1A variant, indicating that the patient is a carrier and NDM was not triggered by autosomal recessive inheritance. Neither chromosomal microarray (CytoScan Dx assay, Affymetrix, ThermoFisher, Waltham, MA, USA) analysis nor methylation-specific multiple ligation-dependent probe amplification detected relevant copy number changes or loss of methylation of 6q24.
DISCUSSION
In neonates with hyperglycemia, NDM should not be immediately ruled out based on its low incidence rate. Impaired glucose homeostasis in the NICU can have numerous causes, including sepsis, stress, early protein intake, and use of various drugs [7]. Limited insulin secretion capacity, increased secretion of catecholamines induced by persistent hypoxia, and absence or delayed advancement of enteral feeding contribute to hyperglycemia and commonly occur in very preterm infants [8]. Hyperglycemia in newborns normally occurs in the first 3 to 5 days after birth, and resolves within 2 or 3 days of onset; however, it may persist up to 10 days after birth [1]. Although the clinical manifestations of dysglycemia in infants could be diverse, the major indication in our patient was shortness of breath, which necessitated the measurement of blood glucose level. Typical clinical signs such as polyuria, polydipsia, weight loss, or growth retardation may not always manifest in the presence of hyperglycemia. Therefore, it is crucial to examine the blood glucose level of any newborn who is acutely ill. As the level of glycated hemoglobin could remain low until the age of 6 months (when hemoglobin A begins to replace fetal hemoglobin), it is not useful as a marker in the assessment of infantile NDM [9].
Both CGM and CSII are products of advanced technology for managing diabetes in pediatric patients. CSII is the subcutaneous continuous administration of short-acting insulin and can safely deliver small dosages even on random feeding. It rapidly switches off when the risk of hypoglycemic episodes is anticipated. Nevertheless, inconsistencies of appetite and feeding schedule and frequent infection of the needle insertion sites are factors that pose difficulties in treating diabetes during the neonatal period [10]. Current CGM devices measure the glucose level in interstitial fluid using an amperometric glucose oxidase needle sensor inserted into the patient’s subcutaneous tissue [11]. Although sensor glucose levels may substantially deviate from the serum levels, and evidence remains unavailable for the clinical benefit and outcome of using CGM devices in the neonatal group, CGM is the only practical option to reduce the stress associated with frequent blood collection. Decreasing the frequency of heel pricks and prompt sensors enabling alarms for hypoglycemic or hyperglycemic episodes allowed for a more effective management of our patient’s blood glucose level by the parents and caregivers, thereby relieving the stress of glucose monitoring. The ideal insertion site for CGM devices is either the thigh or the upper buttock area with minimal subcutaneous fat [1]. Managing blood glucose levels in breastfeeding infants is especially challenging owing to frequent oral intake in variable quantities and repetitive preprandial delivery of small amounts of short-acting insulin.
In addition to CGM, although early insulin therapy at an initial dosage ranging from 0.02 to 0.05 units/kg/hr is widely used, the dosing should be adjusted based on the discretion of the attending clinician [1]. To minimize hypoglycemic episodes, current technology offers a minimally invasive real-time monitoring of blood glucose and continuous delivery of insulin at a rate of as low as 0.025 units/hr. However, its long-term clinical efficacy and safety should be investigated in future studies. A potential limitation of the present case is that the recurrence of diabetes remains unknown, as long-term follow-up is not yet available.
Similar to maturity-onset diabetes of the young, NDM is typically known as a monogenic form of diabetes with highly penetrant inherited mutations in a single gene associated with β-cell function. Advancements in molecular technology have led to the identification of 80% of the etiologic genes of NDM [1]. Nevertheless, the underlying genetic mechanism responsible for NDM, the basic physiology of glucose homeostasis, and the individual roles and interactions of each gene are yet to be fully understood. Recent studies have reported NDM as a polygenic disease with polygenic inheritance, and a diagnosis of diabetes in a child whose parents carry the mutations should not be excluded [12]. Our patient presented with an overt clinical course of TNDM that required insulin therapy for slightly longer than 3 months.
Among the subgroups of NDM, TNDM is characterized by self-limited hyperglycemia, which lasts for a median of 12 weeks and completely resolves by the age of 18 months [13,14]. TNDM accounts for approximately 45% of all NDM cases, PNDM accounts for most of the remaining 55%, and syndromic NDM constitutes <10% [10,15]. Overexpression of the imprinted genes at chromosome 6q24 as well as mutations in ZFP57 and HNF1B have been reported to be responsible for TNDM. Defects in KCNJ11, ABCC8, INS, and SLC2A2 can trigger either TNDM or PNDM [1,16]. Despite the ultimate cessation of exogenous insulin treatment in TNDM, the probability rate of relapse and progression to PNDM is as high as 50% [3]. Intrauterine growth retardation is commonly observed in cases of TNDM because insulin acts as a fetal growth hormone, and, consequently, its insufficiency coupled with failure of transplacental insulin delivery causes newborns with TNDM to be born small for gestational age [17,18].
EIF2AK3, which is found on chromosome 2p11.2, encodes a protein kinase R-like endoplasmic reticulum kinase that is responsible for β-cell development [19]. Its homozygous mutation is a common cause of Wolcott-Rallison syndrome, with clinical features including PNDM, multiple epiphyseal dysphasia, and growth retardation. In contrast, PTF1A, which is found on chromosome 10p12.2, encodes the PTF1A protein involved in pancreatic development. Its coding mutations cause agenesis of the pancreas, resulting in insufficient secretion of insulin, thereby causing diabetes mellitus in early postnatal life [20]. Both EIF2AK3 and PTF1A mutations are associated with PNDM and spontaneously arise with autosomal recessive inheritance [1]. Our patient showed no clinical features associated with either Wolcott-Rallison syndrome or pancreatic agenesis, and neither of his parents were diabetic. Consequently, the patient is expected to be a heterozygous carrier, indicating that the identified EIF2AK3 and PTF1A variants were possibly not related to NDM pathogenicity. Nevertheless, it should be highlighted that various factors, including intrauterine environment, perinatal stress, and diverse genetic background, could cause TNDM and require prompt management. Drawing a solid conclusion based on the genetic alterations identified in our patient was difficult because the pathogenicity is VUS limited and other causative genes of TNDM were not evaluated. When a variant is novel, poorly characterized, or controversial in terms of pathogenicity, it is classified as VUS, and VUS should not be used in clinical decision making because it provides insufficient information for determining pathogenicity [6,21]. Therefore, additional functional studies and wider genetic screening would allow better interpretation and clarification of the effect of the identified variants on our patient.
Lastly, switching from insulin to oral SU was a substantial alternative even if the patient’s glycemic control was deemed unfavorable during the transition period. Oral SU therapy permits insulin secretion through an ATP-independent closure of overly active mutated K-ATP channels. Thus, SU can rapidly ameliorate hyperglycemia in K-ATP channel-associated NDM, which accounts for >50% of all cases of NDM [1]. Early SU therapy may improve speech, motor, and cognitive disabilities, and recent studies have identified a higher prevalence of neurodevelopmental disorders in particular genotypes, although the correlation of the genotypes with neurodevelopmental impairment along with underlying pathophysiology remains to be characterized [22,23]. Although variations of genes encoding the K-ATP channel are known to be the most common and second most common causes of PNDM and TNDM, respectively, genetic confirmation requires a considerable amount of time; however, recent literature suggests considering early SU therapy before a genetic diagnosis is made, to avoid the potential adverse effects of delayed therapy [1,24]. The poor response to SU lowered the possibility of our patient having K-ATP channel mutation-related NDM.
In conclusion, our case suggests that consistent monitoring of blood glucose and genetic testing should be promptly performed in any newborns with persistent hyperglycemia, to prevent further diabetic complications. To date, CSII with CGM is the most effective and practical management for NDM. As the underlying causes of NDM are not fully understood, functional studies and wider genetic screening are needed for identifying and better understanding NDM-causing variants.