INTRODUCTION
Developmental and epileptic encephalopathies (DEE) are a group of progressive early onset epilepsies derived from genetic etiologies and characterized by developmental delay and cognitive deterioration [1]. Genetic epilepsy should be considered as a differential diagnosis when a patient with neonatal seizures does not have a clear history of hypoxic ischemic encephalopathy, vascular events, or infection.
The development of gene sequencing techniques has led to an increasing identification of genetic causes for DEE [2]. Targeted gene panel testing revealed a diagnostic yield of 42.9% and 65.2 % in recent studies from South Korea and the United States, respectively [1,3]. Mutations in KCNQ2 are frequently identified in patients with DEE [1,3] are located on the chromosome (20q13.33) encoding the voltage-dependent potassium channel subunit 7.2 (Kv7.2), and manifest as a broad spectrum of neonatal-onset epilepsy with a varying severity from benign familial neonatal seizures to KCNQ2 encephalopathy [4-6].
KCNQ2 encephalopathy is associated with refractory seizures resulting in a poor neurodevelopmental outcome. Many patients with DEE have seizures refractory to multiple antiepileptic drugs, while those with KCNQ2 encephalopathy reportedly have a positive response to sodium-channel blockers [3,7,8]. In other words, the early identification of KCNQ2 encephalopathy makes it a more direct therapeutic approach possible. Recently, KCNQ2 encephalopathy has been reported to have a characteristic amplitude-integrated electroencephalographic (aEEG) pattern [9]. If there are characteristic findings on the aEEG, this distinct ictal pattern is also expected to be present on conventional electroencephalography (EEG). Early recognition of the electroclinical phenotype with aEEG monitoring can direct genetic testing and a precise therapeutic approach using sodium-channel blockers.
This report describes a case of KCNQ2 encephalopathy with the characteristic ictal aEEG pattern, which was confirmed on conventional EEG.
CASE REPORT
A 3-day-old female infant presented with repetitive myoclonic jerking movements in both her legs that had begun on the day. She was born via spontaneous vaginal delivery at a local hospital at a gestational age of 38 and 4/7 weeks, with a birth weight of 3.5 kg and no perinatal complications. There was no family history of epilepsy or developmental delay. Her spontaneous activities were decreased, but there were no other abnormalities (such as abnormal morphologic features) on physical examination. The results of her laboratory investigations, including metabolic profiles such as serum amino acids and urine organic acids, workup for infections and cerebrospinal fluid study were within normal ranges. Brain magnetic resonance imaging only revealed a low-grade cystic germinal matrix hemorrhage at the left caudothalamic groove (Figure 1).
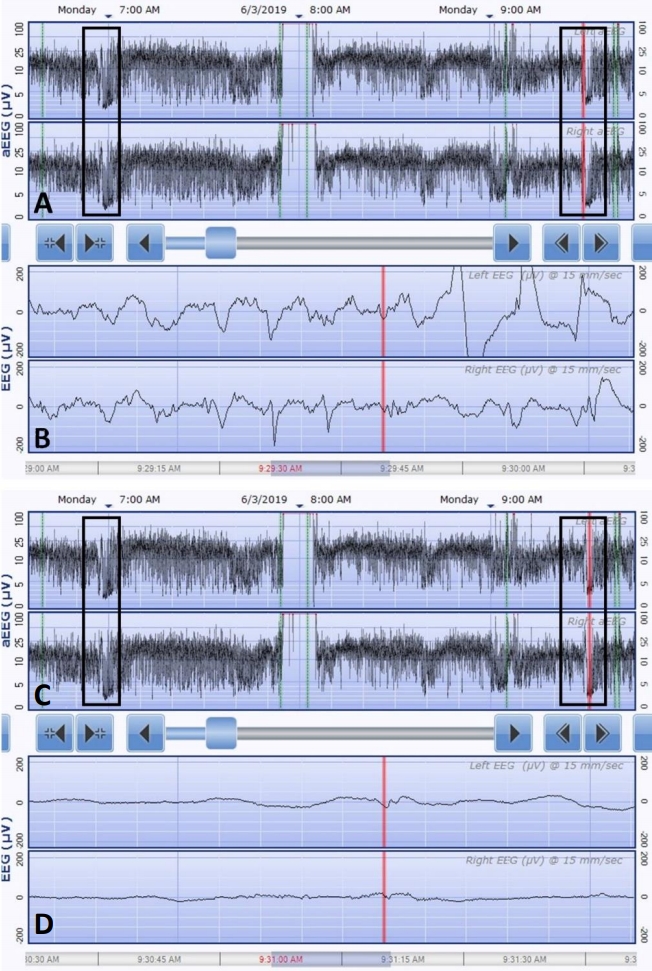
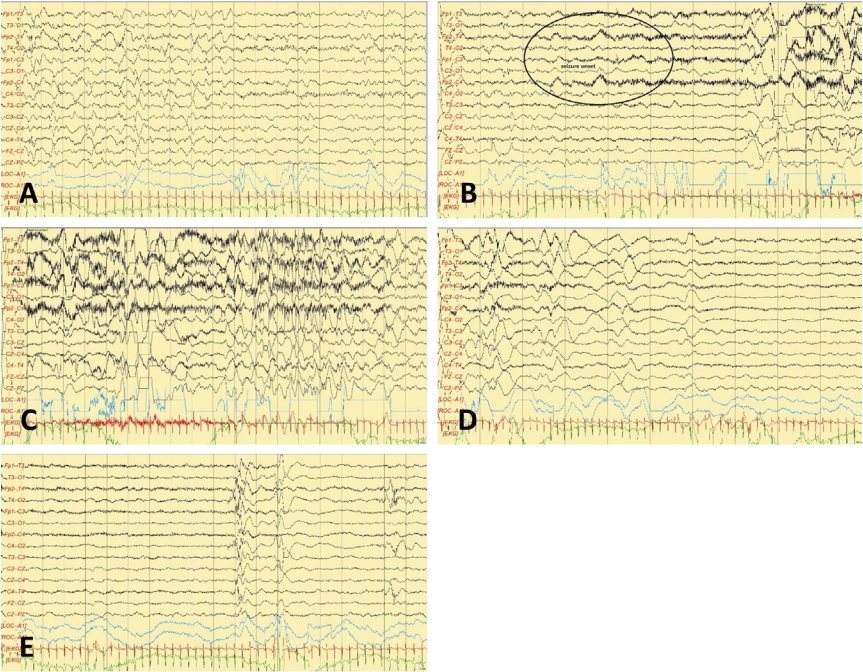
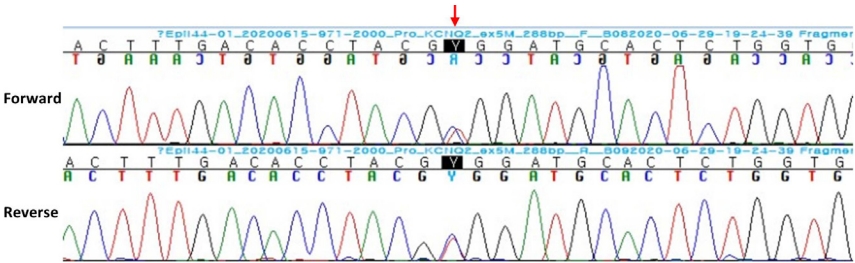
In the neonatal intensive care unit (NICU), seizure monitoring was done using a continuous two-channel aEEG. The seizures lasted for a few minutes and were characterized by tonic posturing followed by unilateral or bilateral clonic jerking. They were always accompanied by apnea and desaturation, and sometimes preceded by crying. Ictal aEEG showed a sudden rise of the highest and lowest margins followed by burst suppression (Figure 2). Conventional EEG showed very frequent episodes of spike or sharp wave discharges, occasionally with a burst suppression pattern. Ictal EEG also showed that the fast activities began from the right frontal area (Figure 3). Multiple antiepileptic drugs (phenobarbital, levetiracetam, topiramate, and phenytoin) were administered; however, seizures and the characteristic ictal aEEG patterns were detected almost every day for over a month. The result of a chromosomal microarray, which was obtained approximately four weeks after testing, was normal. Genetic testing using next generation sequencing also was performed in the 6th week of life and a KCNQ2 mutation (c.794C>T; p.[Ala265Val]) was detected (Figure 4). Consequently, she was diagnosed with KCNQ2 encephalopathy.
At the age of 5 months, the patient continued to be seizurefree, and was being administered levetiracetam, topiramate, lamotrigine, and phenobarbital. She has been undergoing rehabilitation therapy and regular follow-up with developmental tests. A recent Denver Developmental Screening Test revealed a global developmental delay, and the specific delayed age in different domains noted at 12 months of age were: gross motor (7 months of age), fine motor (8 months of age), language (7 months of age), and psychosocial domains (8 months of age).
DISCUSSION
KCNQ2 encephalopathy is associated with a severe form of epilepsy characterized by recurrent tonic seizures with ictal apnea and desaturation, often followed by clonic movements, in the first week of life [7,9,10]. The seizures are extremely resistant to antiepileptic therapy resulting in severe developmental delay, regression, and various comorbidities.
In previous studies, sodium-channel blockers, such as carbamazepine and phenytoin, were reported to be effective in patients with KCNQ2 encephalopathy [3,7,8,11]. Phenobarbital is the most commonly used antiepileptic drug for neonatal seizures, while carbamazepine is rarely used in neonates. Thus, an early diagnosis of KCNQ2 encephalopathy allows more precise management, resulting in more favorable seizure and developmental outcomes. Identifying the etiology of neonatal seizures is crucial for diagnosing and managing seizures. Voltage-gated sodium channels and KCNQ2 potassium channels co-localize and modulate each other at the neuronal complex comprising the channels [12,13]. This structure-function may explain the effectiveness of sodium-channel blockers in patients with KCNQ2 encephalopathy [13].
Recently, a distinctive ictal pattern was reported during aEEG monitoring in infants with KCNQ2 encephalopathy [9]. Continuous aEEG monitoring is widely used for the diagnosis of seizures in real time in the NICU [14]. Our patient also exhibited repeated seizures with the characteristic aEEG findings that have been described by Vilan et al. [9]. They described that the aEEG patterns consist of an abrupt and short rise of the lower and upper margins followed by sudden depression of the background activities. They highlighted the importance of a distinctive aEEG pattern recognition for the genetic diagnosis and targeted treatment of this disorder. Our patient also demonstrated this distinctive aEEG pattern on brain monitoring when she experienced tonic or clonic seizures with apnea or desaturation. Moreover, her seizures were vague, and she mainly experienced desaturation and apnea. She had persistent seizures despite the administration of multiple antiepileptic drugs, indicating epileptic encephalopathy; therefore, we conducted gene panel testing. Identifying this characteristic aEEG pattern of KCNQ2 encephalopathy can lead to early diagnosis and a trial of sodium-channel blockers such as carbamazepine in this condition. Thus, it could influence the selection of an appropriate antiepileptic drug.
This ictal pattern was also confirmed on conventional EEG (Figure 3). Episodes of apnea and desaturation are often underrecognized or misinterpreted as seizures by medical professionals. Therefore, the recognition of this characteristic aEEG or EEG pattern by medical professionals can increase their likelihood of detecting seizures. Early recognition of this electroclinical phenotype by a neonatologist (aEEG) or a neurologist (EEG) can result in genetic testing, leading to the early diagnosis and effective treatment of KCNQ2 encephalopathy with carbamazepine.
Identifying the characteristic ictal pattern on aEEG monitoring facilitates the early detection of KCNQ2 encephalopathy and timely treatment of seizures in patients with refractory seizures. Performing a targeted gene panel after recognizing the characteristic electroclinical phenotype may result in earlier diagnosis and more specific therapeutic management, thus contributing to an improved disease prognosis.