Novel Mutation of SLC26A3 Gene Observed in Congenital Chloride Diarrhea
Article information

Abstract
Congenital chloride diarrhea (CLD) is a rare autosomal recessive disease caused by mutations in the solute carrier family 26 member 3 (SLC26A3) gene on chromosome 7q31. Affected neonates are vulnerable to dehydration, electrolyte imbalance in the form of hyponatremia, metabolic alkalosis, failure to thrive, or even death if left untreated. Genetic testing for mutations should be considered if the clinical diagnosis remains uncertain because early diagnosis and appropriate management are critical to the disease course in CLD. Several mutations have been reported in Korean patients with CLD, with the most common being the c.2063-1G>T mutation. Here, we report the case of a neonate with prenatally suspected CLD with confirmed novel mutations in the SLC26A3 gene (c.2147C>G; p.Ala716Gly).
INTRODUCTION
Congenital chloride diarrhea (CLD) is a rare autosomal recessive disease [1] with approximately 250 cases reported worldwide [2]. Large amounts of watery diarrhea are caused by mutations in the solute carrier family 26 member 3 (SLC26A3) gene on chromosome 7q31, which encodes the intestinal Cl−/HCO3 − exchanger. This transporter is thought to play a role in Cl− absorption and HCO3 − secretion in the terminal ileum and colon; therefore, the mutation results in voluminous osmotic diarrhea with high concentrations of chloride and acidification of the gastrointestinal tract fluids [3,4].
CLD is more prevalent in Poland, Finland, Saudi Arabia, and Kuwait, but has rarely been reported in Korea [5]. Because of this rare prevalence in Korea, it is difficult to consider a differential diagnosis, which can cause a delay in diagnosis and management, worsening the course of the disease. Genetic testing can be useful in the diagnosis of CLD. Several CLD mutations have been reported in Korean patients. Here, we report a case of a neonate prenatally suspected of having a CLD confirmed by genetic testing, leading to the identification of novel mutations in the SLC26A3 gene.
CASE REPORT
A 34-year-old gravida 2, para 1, woman with a history of hypothyroidism and hepatitis B carriers was referred to our department at 26 weeks of gestation. No obvious abnormalities were noted in the first trimester; however, ultrasonography revealed signs of fetal intestinal dilatation at 24 weeks of gestation. Fetal ultrasonography at 26+2 weeks of gestation showed bowel dilatation with multicystic segmentation (Figure 1). Amniocentesis was performed for polyhydramnios, followed by amniotic fluid analysis. The chloride concentration in the amniotic fluid was 111 mEq/L, which was within the upper limit of the normal range [6]. At 31+4 weeks of pregnancy, the mother experienced preterm labor, which was controlled by a tocolytic agent, and was administered a prenatal steroid injection.

Ultrasonographic views at 26 weeks and 2 days of gestation. (A) Bowel dilatation with multicystic segmentation—the honeycomb sign. (B) Polyhydramnios. The length of the single deepest pocket was measured to be 9.57 (>8 cm).
A male neonate weighing 2,800 g was delivered via vacuum-assisted vaginal delivery at 38 weeks of gestation. The Apgar scores were 9 at 1 minute and 10 at 5 minutes and meconium staining was present in the amniotic fluid. Immediately after birth, the neonate was transferred to the neonatal intensive care unit (NICU) because he had a profuse amount of watery stool. He had mild tachycardia (180 to 190 beats per minute) and hypotension (mean arterial pressure, 32 mm Hg) until the day after birth, which was managed with intravenous fluids. Since 270 g (10% of birth weight) was lost by the third day of life, exceeding the physiological weight loss of newborns, mild to moderate dehydration was considered. Neither physical examination nor simple abdominal radiography showed signs of intestinal obstruction; therefore, milk feeding was initiated with partial parenteral nutrition. On the day of birth, electrolytes in the serum and stool were analyzed, and the results were as follows: serum sodium 135 mEq/L, serum potassium 4.6 mEq/L, serum chloride 107 mEq/L, stool sodium 127 mEq/L, stool potassium 10.2 mEq/L, and stool chloride, 136 mEq/L. As CLD was suspected clinically based on the high fecal chloride concentration (>90 mmol) and prenatal ultrasound findings, we analyzed mutations in the SLC26A3 gene to confirm the diagnosis.
After obtaining informed consent, genetic testing was performed at the Department of Laboratory Medicine and Genetics, Samsung Medical Center. Genomic deoxyribonucleic acid (DNA) was isolated from the leukocytes of the patient's peripheral blood, and the sequences of all coding exons and adjacent intron sites of the SCL26A3 gene (reference cDNA sequence: NM_000111.2) were identified using direct sequencing method. Mutation results were reported based on scoring rules for variant classification according to the 2015 American College of Medical Genetics/Association for Molecular Pathology guidelines. Two different pathogenic heterozygous mutations were identified in the SLC26A3 gene of the patient (Figure 2, chromatogram). One was a splice site mutation that was a G to T substitution at the splice acceptor site of intron 18 (c.2063-1G>T), which has been reported in Korean patients [7]. The other was a missense mutation that was a C to G substitution at nucleotide position 2147, leading to the conversion of the 716th amino acid, alanine, to glycine (c.2147C> G; p. Ala716Gly). However, this novel variant has not been previously reported. The parents of the patients were not consanguineous; therefore, we recommended genetic testing of the family, but this recommendation was rejected.
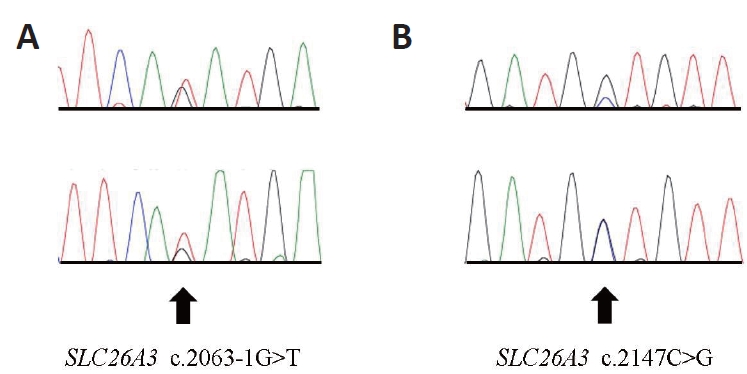
(A, B) Chromatogram of the solute carrier family 26 member 3 (SLC26A3) gene of the patient. Two different pathogenic mutations in heterozygous state were identified. Arrows indicate the variant sequences.
The neonate had watery diarrhea five to six times per day, which lasted until the 4th day after birth. In a follow-up test of electrolytes in the stool, the chloride concentration in the stool decreased to the normal range (Table 1). No signs of dehydration, electrolyte imbalance, or metabolic alkalosis were observed. Symptoms improved from the 5th day of birth, and the patient showed adequate weight gain after milk feeding and was discharged from the NICU on the 13th day of life.

Electrolyte Levels in the Stool of the Patient
DISCUSSION
CLD is a rare autosomal recessive disease characterized by voluminous, watery diarrhea that begins soon after birth and contains a large excess of chloride compared to sodium. A mutation of the SLC26A3 gene, which encodes the apical epithelial Cl–⁄ HCO3– exchanger on chromosome 7q31, causes the disease. This mutation causes impaired intestinal absorption of Cl– and secretion of HCO3 –, as well as secondary inhibition of Na+/H+ exchangers. Consequently, failure to absorb NaCl and fluid from the intestines occurs and manifests clinically as watery diarrhea. Although there are various mutations in SLC26A3, no difference has been observed in their phenotype [8]. In untreated or poorly treated patients with CLD, life-threatening complications, such as severe dehydration and electrolyte imbalance, may occur. Most patients have a favorable prognosis with spontaneous improvement of diarrhea symptoms; however, in some cases, long-term complications such as failure to thrive, male subfertility, psychomotor delay, and rarely, renal involvement, which can eventually lead to end-stage renal disease, may occur [9,10].
As diarrhea begins before birth, it exhibits characteristic prenatal ultrasonography findings. Multiple fluid-filled intestinal loops are observed in a normal-sized stomach. Additionally, marked-tomoderate polyhydramnios is observed during the third trimester of pregnancy [11]. These typical features of the prenatal period may facilitate the prenatal diagnosis of CLD; however, they are not pathognomonic and can be confused with intestinal obstruction, leading to unnecessary surgery after birth [12]. Furthermore, owing to the low incidence and unconventional examination of fecal electrolytes, CLD is easily misdiagnosed. In the current case, the patient reported was quickly diagnosed based on prenatal findings. Another study found a high chloride concentration in the amniotic fluid, which helped diagnose CLD [13]; however, the chloride concentration was at the normal upper limit in our case. If a neonate has prenatal findings of dilated bowel loops and polyhydramnios but no evidence of intestinal obstruction after birth, CLD should be considered, and fecal electrolytes should be evaluated. Diarrhea in neonates can be caused by heterogeneous etiologies, including common acquired etiologies such as allergy or infection [14]. If diarrhea symptoms appear immediately after birth or chronic diarrhea persists, with or without prenatal information, stool electrolytes can be helpful in differentiating the diagnosis.
CLD was first described by Gamble et al. [15] and Darrow [16] in 1945, and approximately 250 cases have since been reported worldwide. Regional differences are observed in its prevalence, with Finland (1:10,000), Poland (1:200,000), and Persian Gulf countries, including Saudi Arabia and Kuwait (1:5,500), showing higher rates than the global average rates because of high rates of consanguinity or founder mutations [5]. CLD is rare in other countries, including Korea, where only a few cases have been reported. The first two cases in Korea were reported by Lee et al. [17] in 1988. Since then, only a few cases have been reported, and mutational analysis was performed for the first time in Korea in 2012 [7]. Several mutations have been reported in Korean patients with CLD; the most common mutation is c.2063-1G>T [18]. Our diagnosis was also confirmed by genetic testing, which revealed novel mutations in the SLC26A3 gene (c.2147C>G; p.Ala716Gly).
In conclusion, early diagnosis and appropriate management are critical to arresting the disease course of CLD but are difficult to consider, particularly in countries with a low prevalence of CLD, such as Korea. Therefore, genetic testing for mutations should be considered to differentiate CLD from other diseases and accurately diagnose CLD, especially if the clinical diagnosis remains uncertain. As more CLD mutations are discovered, a better understanding of the disease will emerge.
Notes
Ethical statement
This study was conducted with the approval from the Institutional Review Board of the Chung-Ang University Hospital (IRB No: 2201-013-19401). This study was exempted from consent from the IRB.
Conflicts of interest
Na Mi Lee is an editorial board member of the journal, but he was not involved in the peer reviewer selection, evaluation, or decision process of this article. No other potential conflicts of interest relevant to this article were reported.
Author contributions
Conception or design: J.H.C., N.M.L.
Acquisition, analysis, or interpretation of data: J.H.C., N.L.Y., N.M.L.
Drafting the work or revising: J.H.C., N.L.Y.
Final approval of the manuscript: All authors read and approved the final manuscript.
Funding
None
Acknowledgements
We thank our colleagues at Chung-Ang University for their helpful feedback and support.