Congenital Long QT Syndrome Type 8 Characterized by Fetal Onset of Bradycardia and 2:1 Atrioventricular Block
Article information

Abstract
An important, albeit rare, cause of fetal bradycardia is long QT syndrome (LQTS). Congenital LQTS is an ion channelopathy caused by mutations in genes encoding cardiac ion channel proteins. Fetal onset of LQTS imposes high risk of life-threatening tachyarrhythmias and sudden cardiac death. Here, we report the case of a female newborn with fetal onset of bradycardia and a 2:1 atrioventricular (AV) block. After birth, a 12-lead electrocardiogram (ECG) revealed bradycardia with QT prolongation of a corrected QT (QTc) interval of 680 ms and pseudo 2:1 AV block. Genetic testing identified a heterozygous Gly402Ser (c.1204G>A) mutation in CACNA1C, confirming the diagnosis of LQTS type 8 (LQT8). The patient received propranolol at a daily dose of 2 mg/kg. Mexiletine was subsequently administered owing to the sustained prolongation of the QT interval and pseudo 2:1 AV block. One week after mexiletine inception, the ECG still showed QT interval prolongation (QTc, 632 ms), but no AV block was observed. There were no life-threatening tachyarrhythmias in a follow-up period of 13 months.
INTRODUCTION
Fetal bradycardia is characterized by a ventricular heart rate slower than 100 beats per minute (bpm), mainly owing to atrioventricular (AV) block. The most common causes of fetal bradycardia are congenital heart diseases. The other important, albeit rare, cause is long QT syndrome (LQTS) [1]. Fetal echocardiography is a useful diagnostic modality for prenatal arrhythmia, and an M-mode trace of ventricular and atrial motion demonstrates fetal cardiac rhythm [2].
LQTS is a primary cardiac channelopathy, characterized by QT prolongation, syncope, and ventricular arrhythmias poses a high risk of sudden cardiac death. LQTS is classified into 17 types, and its causative genes are inherited in an autosomal-dominant or autosomal-recessive form. Mutations of KCNQ1, KCNH2, and SCN5A are the major causes of LQTS [3,4]. In 2004, a G406R mutation in calcium voltage-gated channel subunit α1 (CACNA1C) gene was identified in patients with Timothy syndrome (TS) or LQTS type 8 (LQT8), a specific subtype of LQTS. LQT8 and TS are related to mutations in CACNA1C. TS is a multisystem syndrome associated with QT prolongation and ventricular tachyarrhythmias [5]. Recent advances in genetic analyses have encouraged studies concerning LQT8 [6-8].
CASE REPORT
A 38-year-old woman (gravida 4, para 2) at 34 weeks of gestation was referred to our hospital because of fetal bradycardia. The fetal M-mode echocardiogram revealed bradycardia and a 2:1 AV block with an atrial rate marching out regularly at 132 bpm, and a ventricular rate of 63 bpm (Figure 1). The fetal cardiac structures were normal and the ventricular function was good. She had no medication history during pregnancy or family history suggestive of arrhythmia. Her laboratory tests, including thyroid function tests, electrolytes, and 25-OH vitamin D levels, were all normal.

Fetal M-mode echocardiogram showed a 2:1 atrioventricular block. The atrial rate (A) was 132 bpm and the ventricular rate (B) was 63 bpm.
The infant was delivered by elective cesarean section at 38+2 weeks of gestation and had a birth weight of 2,680 g. The Apgar scores were 8 at 1 minute and 9 at 5 minutes. The 12-lead electrocardiogram (ECG) performed immediately after birth revealed bradycardia, a heart rate of 68 bpm with QT prolongation of a corrected QT (QTc) interval of 680 ms (using Bazett formula), and a pseudo 2:1 AV block (Figure 2). She had no anomalies upon physical examination and echocardiography.
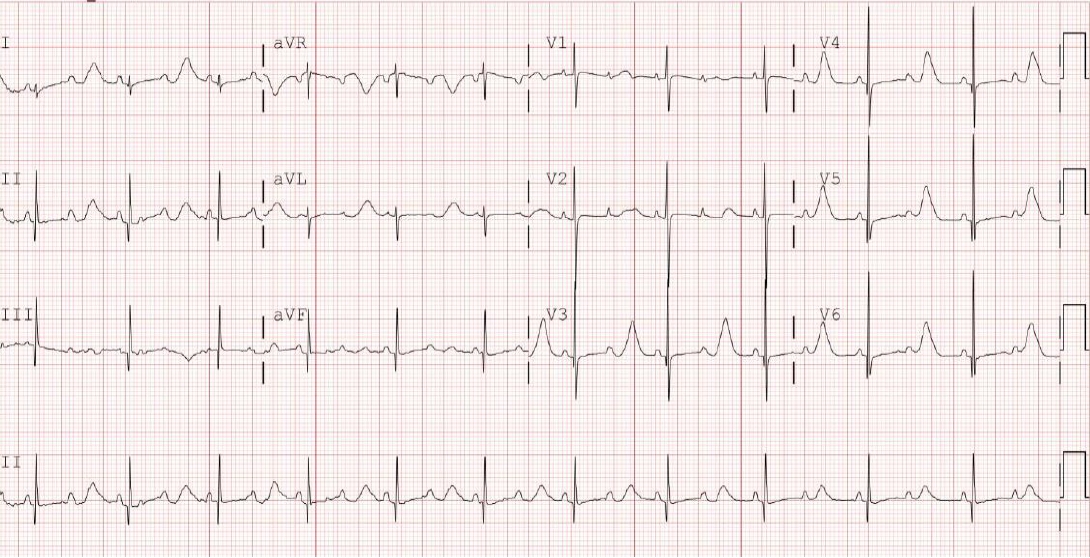
Postnatal 12-lead electrocardiogram showed bradycardia. The heart rate was 68 beats/min with QT prolongation of a corrected QT interval (QTc) of 680 ms (using Bazett formula) and a pseudo 2:1 atrioventricular block.
The baby received propranolol at a daily dose of 2 mg/kg. On the 9th hospital day, mexiletine (6 mg/kg/day) was orally administered because of sustained prolongation of the QT interval and 2:1 AV block. The dose of mexiletine was gradually increased to 15 mg/kg/day. One week after mexiletine inception, the ECG still showed QT interval prolongation (QTc, 632 ms), but no AV block was observed (Figure 3).
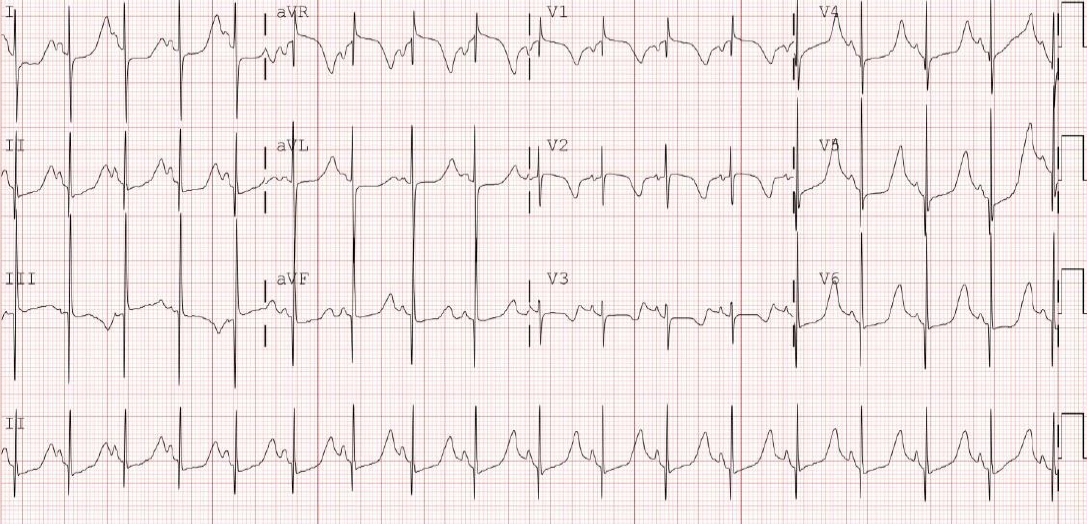
Electrocardiogram continued to show QT interval prolongation (QTc 632 ms) but revealed no atrioventricular block after persistent mexiletine administration.
The baby’s pedigree indicated no cardiac events or sudden death. ECG of the parents and older sister revealed a normal QT interval (QTc, 400 to 430 ms). Genetic testing identified a heterozygous Gly402Ser (c.1204G>A) mutation in CACNA1C, confirming the diagnosis of LQT8. Familial genetic screening was rejected by her parents.
The baby was discharged on the 35th hospital day without complications, and continued oral propranolol and mexiletine. No life-threatening tachyarrhythmias were noted during a follow-up period of 13 months. The patient did not develop seizures or neurodevelopmental delays and dysmorphic features.
DISCUSSION
This is the first report describing congenital LQT8 characterized by fetal onset of bradycardia and 2:1 AV block, in Korea. Currently, LQTS is classified into 17 genotypes. Three major genes, namely, KCNQ1, KCNH2, and SCN5A (LQT1 through LQT3), are known to contribute to 75% of all cases. Efforts to explain the remaining heritability of LQTS resulted in the discovery of multiple minor LQTS genes, numbered LQT4 through LQT17 [9]. The minor LQTS genes collectively account for 5% to 10% cases [3], while LQT8 accounts for 1% to 2% of all LQTS cases [7].
Sinus bradycardia is the most common manifestation of fetal LQTS [10]. Although most fetal arrhythmias are benign, some may cause sudden cardiac death. It is important to diagnose arrhythmia through prenatal examination to allow informed choice regarding delivery in a facility that can provide proper post-delivery treatment [1].
Skinner et al. [4] summarized the treatment for LQTS based on severity. Long-acting beta blockers are recommended as the first treatment for all types of LQTS. Left sympathetic denervation has been shown to reduce the occurrence of ventricular tachyarrhythmias in high-risk LQTS. Implanted defibrillators are reserved for those who have a cardiac arrest. Despite continued treatment, life-threatening tachyarrhythmias and sudden cardiac death occurred in many pediatric patients [10,11]. Fortunately, our patient had a stable cardiovascular state. Hence, we considered initiating medical treatment. There were no life-threatening tachyarrhythmias during the follow-up period.
As shown in Figure 2, the ECG revealed extreme QT prolongation (QTc 680 ms) associated with 2:1 AV block. However, PP intervals (440 ms) were much shorter than the prolonged QT interval. If the ventricular refractory period is extremely prolonged, more than the sinus rate, it may present as a pseudo 2:1 AV block. The pseudo 2:1 AV block appears to be unique to congenital LQTS [12]. LQTS with 2:1 AV block poses a high risk of sudden cardiac death [2].
Mazzanti et al. [13] and Okuwaki et al. [14] reported mexiletine to be effective in LQT3 with 2:1 AV block. Mexiletine has the potential to shorten the QT interval by inhibiting the late sodium current. We considered that the administration of oral mexiletine could be helpful in our case, and 1:1 AV conduction was recovered.
In this case, LQT8 was diagnosed with genetic DNA sequencing, Caras genetic analysis suggested an arrhythmia phenotype and consequent clinical outcomes [9]. The CACNA1C gene encodes the core α1 subunit of the voltage-gated L-type calcium channel. In 2004, a CACNA1C mutation was reported in the first description of TS. TS is a rare genetic disorder accompanied by cardiac phenotypes, including QT prolongation, congenital heart disease, and extracardiac phenotypes, such as syndactyly, facial dysmorphism, seizure, and developmental delay [5]. Since then, additional CACNA1C variants have been reported in patients with non-syndromic LQTS, referred to as LQT8 [7]. Our patient did not have any other TS characteristics during the follow-up period. This confirmed the diagnosis of LQT8.
In conclusion, LQTS is highlighted by the fact that it contributes to sudden cardiac death. Delivery should be performed in a center with adequate facilities, and prompt treatments should be adopted to prevent life-threatening arrhythmia after birth [1,2]. To improve the outcome in such cases, an effort to provide more attention to fetal arrhythmia is imperative. Moreover, recent advances in genetic analyses can provide additional information regarding the arrhythmia phenotype and clinical outcomes [9].
Notes
Ethical statement
This study was approved by the Institutional Review Board (IRB) of the Pusan National University Yangsan Hospital (approval number: 05-2020-204). Written informed consent by the patients was waived due to a retrospective nature of our study.
Conflicts of interest
No potential conflict of interest relevant to this article was reported.
Author contributions
Conception or design: D.J., H.D.L., J.H.B.
Acquisition, analysis, or interpretation of data: D.J., T.K., J.H.B.
Drafting the work or revising: D.J., H.K., J.H.B.
Final approval of the manuscript: H.D.L., J.H.B.
Acknowledgements
None