INTRODUCTION
Partial trisomy 1q, a rare chromosomal abnormality, has been reported as either pure trisomy or unbalanced translocation [1]. Partial duplications of chromosome 1q mainly involve duplications in the 1q32qter region [2-4]. Most infants with this chromosomal abnormality are stillborn or die prematurely. If they survive, severe psychomotor and mental development delays usually occur [1,4,5]. Patients diagnosed postnatally typically die due to respiratory or hepatic failure. However, to our knowledge, treatment modalities for mental or developmental delays in such cases have not yet been reported. In this report, we describe an 18-month-old patient with partial duplication of the 1q32.1-44 segment who exhibited improvement in the developmental status with rehabilitation.
CASE REPORT
A 36-year-old gravida 3, para 1 female with no specific personal or familial history conceived through in vitro fertilization. Lateral ventricular dilatation in the fetus was detected on prenatal sonography performed at 21 weeks and 4 days of gestation. At 36 weeks of gestation, ultrasonography revealed subgaleal fluid collection, and magnetic resonance imaging (MRI) showed extensive atrophy of the left cerebral hemisphere of the fetus.
The male neonate was born at 37 weeks and 5 days of gestation by cesarean section, and the Apgar score was 4 at 1 minute (color 0, respiration 0, muscle tone 1, reflex 1, heart rate 2) and 6 at 5 minutes (color 1, respiration 1, muscle tone 1, reflex 1, heart rate 2). Positive-pressure ventilation was required immediately after birth. His weight was 3.458 kg (75 to 90 percentile), head circumference was 38 cm (>90 percentile), and length was 48 cm (50 to 75 percentile). Polydactyly of the right thumb, long thin fingers and toes, micrognathia, microphthalmia, macrocephaly, and low-set ears were noted in addition to a heart murmur (Figure 1).
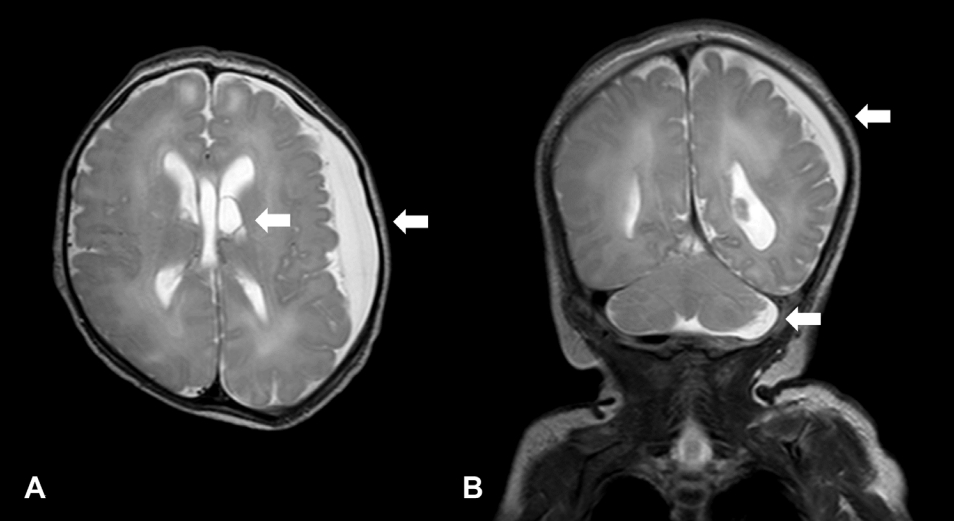
Brain MRI revealed diffuse subdural fluid collection in the left cerebral and cerebellar convexity, a dilated left temporal horn, and choroid plexus cysts in the lateral ventricle during the neonatal period (Figure 2). Echocardiography revealed a patent ductus arteriosus (PDA) and ventricular septal defect (VSD). The patient had symptoms of dyspnea since hospitalization, for which a humidified high-flow nasal cannula was provided. However, after two courses of ibuprofen, dyspnea persisted, the ductus arteriosus did not close, and the patient developed cardiomegaly. PDA ligation was performed at the post-conceptional age of 39 weeks and 4 days despite persistent symptoms of heart failure. Enalapril was prescribed.
Chromosomal analysis revealed a 46,XY,rec(1)dup(1)(q32.1q44) inv(1)(p36.3q32.1)pat karyotype. A paternal chromosomal study confirmed the presence of a 46,XY,inv(1)(p36.3q32.1) karyotype, whereas the maternal chromosomal study was normal (Figure 3).
Breast-feeding progressed smoothly without heart or liver failure in the infant, and his weight gain was as expected. The patient was discharged with instructions for follow-up. Enalapril was discontinued at the age of 5 months. The patient underwent surgery for a VSD at 7 months of age. Additionally, the patient was admitted twice with acute bronchiolitis and hypostatic pneumonia.
A follow-up brain MRI at 17 months showed minimal subdural fluid collection and a decreased volume of the deep cerebral white matter in both the cerebral hemispheres, indicating the progression of secondary ventriculomegaly.
The patientŌĆÖs initial evaluation of developmental status was performed at 12 months of age using the Bayley Scales of Infant and Toddler Development III. All subtests revealed extremely low composite scores across the domains: cognitive, 55; language, 47; motor, 46; and adaptive behavior, 63 (score for the social-emotional aspect was not recorded). The developmental age was assessed to be 1 to 2 months. We used the Gross Motor Function Measure (GMFM) to evaluate the patient's gross motor function accurately. The score was 58.8% for the lying and rolling dimensions, indicating that he could roll over and partially lift his head in the prone position. We initiated rehabilitation at 11 months of age and continued an appropriate rehabilitation program. The program consisted of central nervous system developmental rehabilitaion therapy, occupational therapy, mat and mobility therapy, and daily living movement training therapy conducted in cooperation with other medical centers to facilitate postural control and adaptive responses. A second evaluation performed at 18 months of age revealed minor functional improvements. He scored 25% on the sitting dimension of the GMFM, which indicated the ability to sit with support temporarily. The Bayley Scales also showed slight improvements in gross motor functions, found to be the equivalent to motor development at 6 months of age, and the remaining scores were as follows: cognitive, 55; language, 53; motor, 46; social-emotional, 80; and adaptive behavior, 53.
DISCUSSION
Partial trisomy 1q could include all segments of the q arm of chromosome 1; however, most cases are usually constituted by trisomy 1q32-qter and 1q42-qter [3-6]. Partial trisomy 1q is classified into two groups: with or without another chromosomal aberration (i.e., pure type). A partial 1q trisomy due to unbalanced translocations with accompanying partial monosomies of other chromosomes produces complex malformation patterns because both the participating chromosomes contribute to the phenotype. However, pure partial duplications of the 1q chromosome are helpful for studying and understanding the resultant phenotypic features. Most cases of pure partial chromosome 1q duplications involve duplications in the 1q32qter region [1].
Both de novo and familial chromosomal aberrations have been reported, with the de novo type being more often reported [4,7,8]. Moreover, based on the direction of the duplicated segment, they can be divided into direct and inverted duplications. The type of duplication causes significant differences in the prognosis. For example, inverted duplications have been associated with telomeric deletions [9]. The location and extension of the deleted segment, breakpoints of the inversion, and length of the region included in the trisomy influence the phenotype [3]. Partial trisomy 1q32.1-44 was present in our patient because of a balanced familial rearrangement. The inverted chromosome of the father affected the patientŌĆÖs chromosomes.
WNT genes play important roles in facial and cranial morphogenesis. Among the WNT genes, WNT9A and WNT3A were located within the region of intersection between the duplications on chromosome 1 (1q42). WNT9A is associated with the chondrogenic development of craniofacial structures [10]. Thus, partial duplication of the distal long arm of chromosome 1 presents with similar dysmorphic facial features, including microcephaly, triangular face, microretrognathia, pointed chin, slanted and narrow palpebral fissures, broad and flat nasal bridge, cleft lip and palate, and low-set ears.
In addition to facial deformities, partial trisomy 1q is associated with intrauterine growth retardation, failure to thrive, and developmental delays. Other anomalies (in the cardiovascular, gastrointestinal, and urogenital systems) are related to other chromosomal aberrations. Anomalies in the vital organs markedly increase the risk of death. The most common cause of death in the reported cases was respiratory complications. One patient died of pneumonia at 6 weeks of age, and another died due to pneumonia caused by complications related to tracheostomy insertion for laryngomalacia [8,11]. Another critical event was reported by Sillen et al. [11]. The patient survived, but the bronchopneumonia lasted from 6 months to 5 years of age, necessitating the use of a respirator twice. Severe pneumonia caused a cardiac arrest at 15 months of age. According to reported cases, malformations of the respiratory tract can cause potentially life-threatening complications. Nine of the 13 patients were diagnosed with cardiac diseases, including congenital heart defects such as PDA and patent foramen ovale. Congenital heart diseases are heterogeneous and are primarily attributed to multifactorial inheritance [2]. In the present case, the patient had heart failure due to PDA and a VSD. However, no other organ malformation was observed.
In most cases, pregnancies with a prenatal diagnosis of partial trisomy 1q are likely to be terminated [1,5]. The patients diagnosed postnatally typically died due to numerous complications. Even if these patients survived, they experienced severe psychomotor and mental developmental delays [10,11]. The patient had severe developmental delays in the present case. Therefore, rehabilitation treatment was initiated and continued from the age of 11 months. At 18 months, he was temporarily able to sit without assistance. However, it is difficult to determine whether a patient's developmental state is a result of the natural development over time or an improvement due to rehabilitation. Therefore, more extensive studies are necessary to verify the effectiveness of the rehabilitation treatment.
This case report describes a patient who developed adequate ability to sit without assistance after receiving continuous rehabilitative measures consisting of central nervous system developmental rehabilitation therapy, occupational therapy, mat and mobility therapy, and daily living movement training therapy. Rehabilitation treatment can, therefore, improve the patient's development to varying degrees.